Customizing nf-core Modules: Building a Domain-Specific Variant Calling Library

While nf-core provides a comprehensive, well-maintained library of Nextflow modules for bioinformatics, organizations often need domain-specific variants tailored to their analysis pipelines and computational infrastructure. The gianglabs/nf-modules repository demonstrates how to extend the nf-core framework by integrating both shared nf-core modules and custom organization-specific modules optimized for genomic data processing, particularly variant discovery and annotation workflows. This post explores the core differences between standard nf-core modules and the customized gianglabs implementation, including specialized module variants, domain-focused subworkflows, and adapted CI/CD practices.
After re-organization the modules and subworkflows, the nf-germline-short-read-variant-calling now can integrated with gianglabs/nf-modules
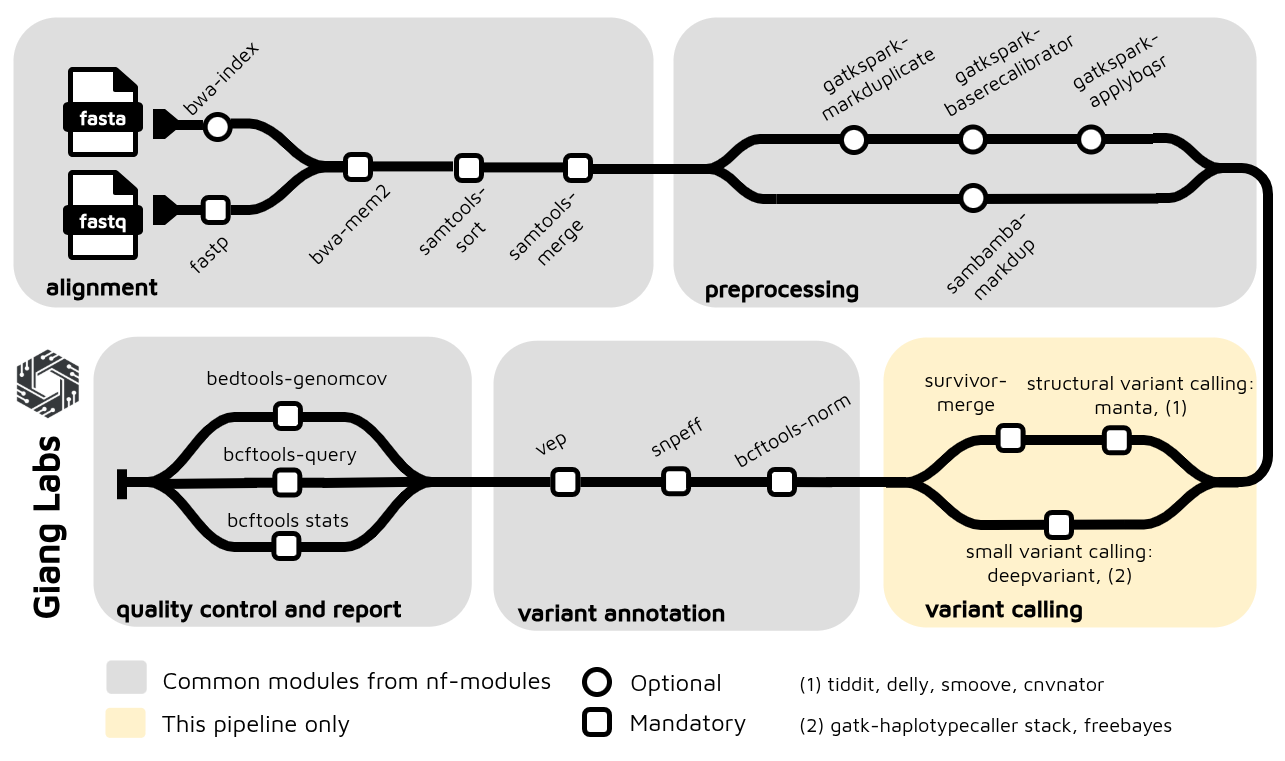
1. Understanding the Differences
1.1. What is nf-core/modules?
The standard nf-core/modules repository provides:
- A curated, community-maintained collection of Nextflow processes
- Modules with standardized structure, testing, and documentation
- Official nf-core-branded modules (e.g.,
bwa/mem,samtools/sort,fastqc) - Shared infrastructure available to all nf-core pipelines
- Strict quality standards and peer review
1.2. Why Create a Custom Organization-Specific Repository?
Custom repositories like gianglabs/nf-modules extend nf-core by adding:
- Domain-specific modules not available in nf-core (e.g., specialized GATK variants)
- Tool variants optimized for your infrastructure (e.g., Spark-based GATK)
- Pre-configured subworkflows combining multiple modules into domain pipelines
- Organizational standardization across pipelines in a research group or company
- Faster iteration on modules without going through community review process
- Cross-organization workflows mixing nf-core and custom modules
1.3. Architecture: Multi-Organization Module Repository
gianglabs/nf-modules/
│
├── modules/
│ ├── nf-core/ # Shared nf-core modules (dependencies)
│ │ ├── fastqc/
│ │ ├── samtools/
│ │ ├── bwa/
│ │ └── ... (installed as dependencies)
│ │
│ └── gianglabs/ # Custom organization-specific modules
│ ├── bwamem2/ # Customized alignment (local optimization)
│ ├── gatk/ # Standard GATK tools
│ ├── gatkspark/ # Spark-based GATK variants (HPC optimized)
│ ├── fastp/ # Customized QC/trimming
│ ├── samtools/ # Custom samtools variants
│ ├── bcftools/ # Variant processing
│ ├── vep/ # Variant annotation
│ ├── snpeff/ # Variant annotation
│ └── ... (other custom modules)
│
├── subworkflows/
│ ├── nf-core/ # Shared nf-core subworkflows
│ │
│ └── gianglabs/ # Domain-focused composite workflows
│ ├── alignment/ # QC → Alignment → Sorting
│ ├── alignment_preprocessing/ # Pre-alignment QC
│ ├── variant_alignment_quality_control/ # BAM validation
│ └── variant_annotation/ # VEP/snpEff annotation
│
└── Pixi + GitHub Actions CI/CD
├── Reusable workflows (detect-changes)
└── Matrix-based testing (modules × subworkflows)
2. Core Differences: Pragmatic Testing Strategy
2.1. nf-core Approach: Comprehensive but Resource-Heavy
The standard nf-core/modules testing philosophy emphasizes broad compatibility:
# nf-core approach (complex)
testing:
- conda environment # Full dependency management
- docker container # Platform-agnostic testing
- singularity image # HPC cluster compatibility
- GPU variant tests # Accelerated compute scenarios
- Multiple shards # Distributed test execution
Requirements:
- Multiple runtime environments configured in CI/CD
- Docker images + Singularity conversions
- GPU runners for GPU tests
- Higher CI/CD cost and complexity
- Longer feedback loops
Problem: Your HPC environment doesn't use GPU, and maintaining conda + docker + singularity testing adds complexity without benefit for your specific use case.
2.2. gianglabs Pragmatic Approach: Focused on Reality
The customized gianglabs/nf-modules testing strategy is environment-aware and cost-conscious:
# gianglabs approach (focused)
Testing Strategy:
GitHub Actions CI/CD:
- docker container only # Fast, resource-efficient validation
- CPU tests only # Matches actual HPC production
- Quick feedback (<5 min) # Faster iteration
Local HPC Development:
- singularity/apptainer # Native HPC runtime
- Manual testing before push # Comprehensive validation
- Real-world execution profile # True performance testing
Key Philosophy:
- Docker for CI/CD validation: Quickly catch obvious issues (syntax, imports, basic logic)
- Singularity for real testing: Run on actual HPC environment with actual data patterns
- Single source of truth: One Docker container that converts to Singularity
- No redundant multi-environment testing: If Docker→Singularity conversion fails, it's a container issue (not a test design issue)
2.3. Practical Benefits
Before (nf-core style):
Pull Request
├─ Test conda env (10 min) → May pass but conda not used in production
├─ Test docker (10 min) → May pass but HPC uses singularity
├─ Test singularity (15 min) → HPC-relevant but slow
├─ Test GPU (20 min) → Unnecessary for CPU-only HPC
└─ Total CI time: ~45 min (wasted on irrelevant scenarios)
After (gianglabs pragmatic):
Pull Request
├─ Test docker (10 min) → Validates code quickly
└─ Total CI time: ~10 min (focused feedback)
Developer (Local HPC):
├─ Manual singularity test → Comprehensive validation
├─ Test with actual data → Real-world performance
├─ Push when confident
└─ CI catches edge cases in 10 min
Savings:
- Reduced GitHub Actions costs
- Focus on actual execution environment (Singularity on HPC)
- Docker→Singularity conversion is a container concern, not a test design concern
2.4. Module Container Specification
To maintain consistency across Docker and Singularity environments, the gianglabs approach uses a single Docker image as the source of truth. Nextflow automatically converts this image to Singularity at runtime, eliminating the need for dual container definitions.
Simple container specification:
// Module specifies ONLY Docker - Nextflow handles Singularity conversion
process EXAMPLEMODULE {
container "quay.io/biocontainers/bwamem2:2.0--h1296002_0"
script:
"""
bwa-mem2 mem ...
"""
}
This pragmatic approach contrasts with nf-core's fastp module, which maintains complex conditional logic for multiple container engines:
process FASTP {
tag "$meta.id"
label 'process_medium'
conda "${moduleDir}/environment.yml"
container "${ workflow.containerEngine == 'singularity' && !task.ext.singularity_pull_docker_container ?
'https://community-cr-prod.seqera.io/docker/registry/v2/blobs/sha256/55/556474e164daf5a5e218cd5d497681dcba0645047cf24698f88e3e078eacbd09/data' :
'community.wave.seqera.io/library/fastp:1.1.0--08aa7c5662a30d57' }"
input:
tuple val(meta), path(reads), path(adapter_fasta)
val discard_trimmed_pass
val save_trimmed_fail
val save_merged
output:
tuple val(meta), path('*.fastp.fastq.gz') , optional:true, emit: reads
tuple val(meta), path('*.json') , emit: json
tuple val(meta), path('*.html') , emit: html
tuple val(meta), path('*.log') , emit: log
tuple val(meta), path('*.fail.fastq.gz') , optional:true, emit: reads_fail
tuple val(meta), path('*.merged.fastq.gz'), optional:true, emit: reads_merged
tuple val("${task.process}"), val('fastp'), eval('fastp --version 2>&1 | sed -e "s/fastp //g"'), emit: versions_fastp, topic: versions
when:
task.ext.when == null || task.ext.when
script:
def args = task.ext.args ?: ''
def prefix = task.ext.prefix ?: "${meta.id}"
def adapter_list = adapter_fasta ? "--adapter_fasta ${adapter_fasta}" : ""
def fail_fastq = save_trimmed_fail && meta.single_end ? "--failed_out ${prefix}.fail.fastq.gz" : save_trimmed_fail && !meta.single_end ? "--failed_out ${prefix}.paired.fail.fastq.gz --unpaired1 ${prefix}_R1.fail.fastq.gz --unpaired2 ${prefix}_R2.fail.fastq.gz" : ''
def out_fq1 = discard_trimmed_pass ?: ( meta.single_end ? "--out1 ${prefix}.fastp.fastq.gz" : "--out1 ${prefix}_R1.fastp.fastq.gz" )
def out_fq2 = discard_trimmed_pass ?: "--out2 ${prefix}_R2.fastp.fastq.gz"
// Added soft-links to original fastqs for consistent naming in MultiQC
// Use single ended for interleaved. Add --interleaved_in in config.
if ( task.ext.args?.contains('--interleaved_in') ) {
"""
[ ! -f ${prefix}.fastq.gz ] && ln -sf $reads ${prefix}.fastq.gz
fastp \\
--stdout \\
--in1 ${prefix}.fastq.gz \\
--thread $task.cpus \\
--json ${prefix}.fastp.json \\
--html ${prefix}.fastp.html \\
$adapter_list \\
$fail_fastq \\
$args \\
2>| >(tee ${prefix}.fastp.log >&2) \\
| gzip -c > ${prefix}.fastp.fastq.gz
"""
} else if (meta.single_end) {
"""
[ ! -f ${prefix}.fastq.gz ] && ln -sf $reads ${prefix}.fastq.gz
fastp \\
--in1 ${prefix}.fastq.gz \\
$out_fq1 \\
--thread $task.cpus \\
--json ${prefix}.fastp.json \\
--html ${prefix}.fastp.html \\
$adapter_list \\
$fail_fastq \\
$args \\
2>| >(tee ${prefix}.fastp.log >&2)
"""
} else {
def merge_fastq = save_merged ? "-m --merged_out ${prefix}.merged.fastq.gz" : ''
"""
[ ! -f ${prefix}_R1.fastq.gz ] && ln -sf ${reads[0]} ${prefix}_R1.fastq.gz
[ ! -f ${prefix}_R2.fastq.gz ] && ln -sf ${reads[1]} ${prefix}_R2.fastq.gz
fastp \\
--in1 ${prefix}_R1.fastq.gz \\
--in2 ${prefix}_R2.fastq.gz \\
$out_fq1 \\
$out_fq2 \\
--json ${prefix}.fastp.json \\
--html ${prefix}.fastp.html \\
$adapter_list \\
$fail_fastq \\
$merge_fastq \\
--thread $task.cpus \\
--detect_adapter_for_pe \\
$args \\
2>| >(tee ${prefix}.fastp.log >&2)
"""
}
stub:
def prefix = task.ext.prefix ?: "${meta.id}"
def is_single_output = task.ext.args?.contains('--interleaved_in') || meta.single_end
def touch_reads = (discard_trimmed_pass) ? "" : (is_single_output) ? "echo '' | gzip > ${prefix}.fastp.fastq.gz" : "echo '' | gzip > ${prefix}_R1.fastp.fastq.gz ; echo '' | gzip > ${prefix}_R2.fastp.fastq.gz"
def touch_merged = (!is_single_output && save_merged) ? "echo '' | gzip > ${prefix}.merged.fastq.gz" : ""
def touch_fail_fastq = (!save_trimmed_fail) ? "" : meta.single_end ? "echo '' | gzip > ${prefix}.fail.fastq.gz" : "echo '' | gzip > ${prefix}.paired.fail.fastq.gz ; echo '' | gzip > ${prefix}_R1.fail.fastq.gz ; echo '' | gzip > ${prefix}_R2.fail.fastq.gz"
"""
$touch_reads
$touch_fail_fastq
$touch_merged
touch "${prefix}.fastp.json"
touch "${prefix}.fastp.html"
touch "${prefix}.fastp.log"
"""
}
gianglabs simplification: Domain-focused modules prioritize clarity and maintainability. For example, while nf-core's fastp supports single-end, paired-end, and interleaved modes, the gianglabs FASTP_TRIM module focuses exclusively on paired-end reads—the production use case:
process FASTP_TRIM {
tag "${meta.id}"
label 'process_medium'
container 'quay.io/biocontainers/fastp:1.1.0--heae3180_0'
input:
tuple val(meta), path(reads)
output:
tuple val(meta), path("*_trimmed_{1,2}.fastq.gz"), emit: reads
tuple val(meta), path("*.html"), emit: html
tuple val(meta), path("*.json"), emit: json
path "versions.yml", emit: versions
when:
task.ext.when == null || task.ext.when
script:
def args = task.ext.args ?: ''
def prefix = task.ext.prefix ?: "${meta.read_group}"
"""
# Adapter trimming, quality filtering, and QC with fastp
fastp \\
--thread ${task.cpus} \\
--in1 ${reads[0]} \\
--in2 ${reads[1]} \\
--out1 ${prefix}_trimmed_1.fastq.gz \\
--out2 ${prefix}_trimmed_2.fastq.gz \\
--html ${prefix}_fastp.html \\
--json ${prefix}_fastp.json \\
--qualified_quality_phred 20 \\
--length_required 50 \\
--detect_adapter_for_pe \\
${args}
# Create versions file
cat <<-END_VERSIONS > versions.yml
"${task.process}":
fastp: \$(fastp --version 2>&1 | grep -oP 'fastp \\K[0-9.]+')
END_VERSIONS
"""
}
3. CI/CD and Core Implementation Details
3.1. Multi-Language Linting Strategy via Makefile and Pixi
Challenge: Bioinformatics modules often combine code from multiple programming languages:
- Nextflow/Groovy: Workflow orchestration
- Python: Data processing, utility scripts
- R: Statistical analysis, visualization
gianglabs solution: Unified linting workflow via Makefile + Pixi environment
Pixi Configuration for Multi-Language Support
# pixi.toml - Reproducible environment with language tools
[dependencies]
python = "3.11.*"
nf-test = ">=0.9.5,<0.10"
nextflow = ">=25.10.4,<26"
ruff = ">=0.1.0" # Python linter + formatter
r = ">=4.0" # R interpreter
r-lintr = ">=3.0.0" # R linter
[pypi-dependencies]
nf-core = ">=3.5.2, <4"
pre-commit = ">=3.0.0"
Benefits:
- All language runtimes in one reproducible environment
- No system dependencies scattered across machines
pixi shellactivates complete dev environment
Makefile: Convenient Linting Commands
# Makefile - Centralized development commands
.PHONY: lint lint-python lint-r lint-nf test clean
# Run all linters
lint: lint-python lint-r pre-commit
# Python linting (Ruff)
lint-python:
@echo "Linting Python files with Ruff..."
ruff check modules/ subworkflows/ --fix
ruff format modules/ subworkflows/
# R linting (lintr)
lint-r:
@echo "Linting R files with lintr..."
Rscript -e "lintr::lint_dir('modules/', recurse = TRUE)"
Rscript -e "lintr::lint_dir('subworkflows/', recurse = TRUE)"
# Pre-commit hooks
pre-commit:
@echo "Running pre-commit checks..."
pre-commit run --all-files
# Run tests
test:
@echo "Running nf-test..."
nf-test test --profile docker
# Clean artifacts
clean:
rm -rf .nf-test work/
Developer Workflow
# Activate Pixi environment (installs Python, R, Nextflow, lintr, ruff)
pixi shell
# Run all linters in one command
make lint
# Or run specific language linters
make lint-python # Check Python code
make lint-r # Check R code
make pre-commit # YAML, Nextflow syntax, JSON schemas
# Run tests
make test
# Clean artifacts
make clean
Language-Specific Tools
Python (Ruff):
# Check imports, undefined variables, and style violations
ruff check modules/gianglabs/ --select I,E1,E4,E7,E9,F,UP,N
# Auto-format code
ruff format modules/gianglabs/
R (lintr):
# Check naming conventions, style, and unused variables
Rscript -e "lintr::lint_dir('modules/gianglabs/', recurse = TRUE)"
Nextflow/Groovy (pre-commit hooks):
# .pre-commit-config.yaml
- repo: https://github.com/pre-commit/mirrors-prettier
hooks:
- id: prettier
files: \.(nf|groovy)$
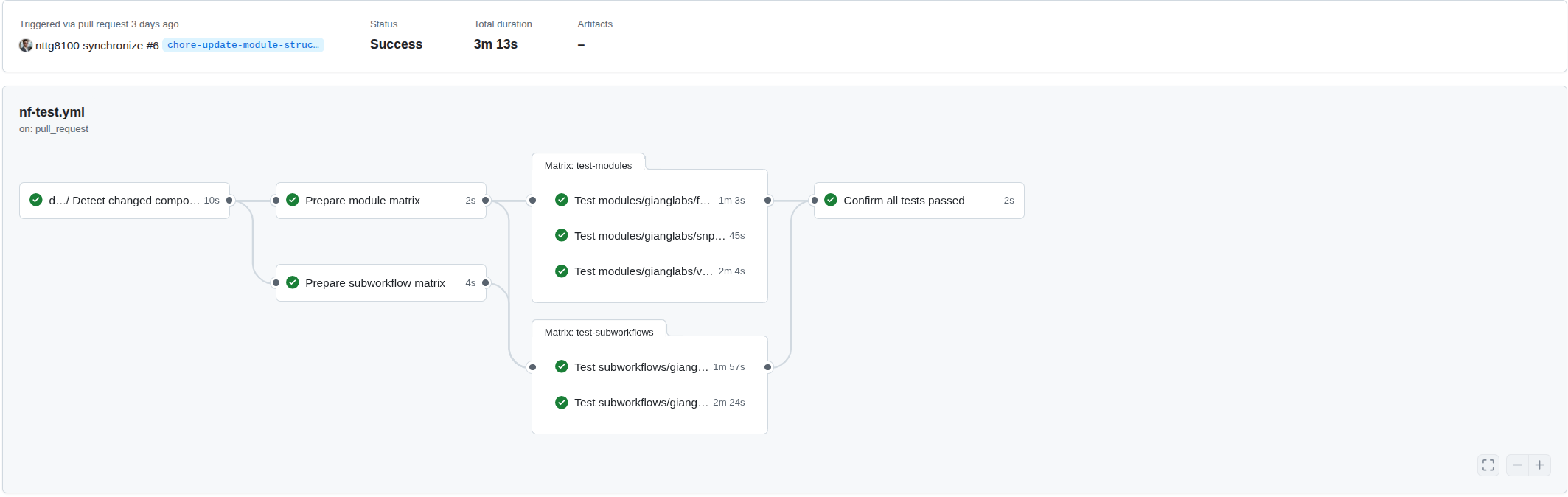
3.2. Reusable Workflow Pattern: detect-changes.yml
To optimize CI/CD efficiency, gianglabs uses a reusable GitHub Actions workflow that detects which modules or subworkflows have changed in a pull request, then triggers tests only for affected components. This approach avoids unnecessary test runs while ensuring comprehensive validation:
# .github/workflows/detect-changes.yml
name: Detect changed components
on:
workflow_call:
outputs:
modules:
description: "Space-separated list of changed modules"
value: ${{ jobs.detect-changes.outputs.modules }}
subworkflows:
description: "Space-separated list of changed subworkflows"
value: ${{ jobs.detect-changes.outputs.subworkflows }}
jobs:
detect-changes:
runs-on: ubuntu-latest
outputs:
modules: ${{ steps.changed-modules.outputs.all_changed_files }}
subworkflows: ${{ steps.changed-subworkflows.outputs.all_changed_files }}
steps:
- name: Get changed modules
uses: tj-actions/changed-files@v46.0.5
with:
dir_names: "true"
dir_names_max_depth: 4
files: modules/gianglabs/**
base_sha: ${{ github.event.pull_request.base.ref || 'origin/main' }}
- name: Get changed subworkflows
uses: tj-actions/changed-files@v46.0.5
with:
dir_names: "true"
dir_names_max_depth: 3
files: subworkflows/gianglabs/**
base_sha: ${{ github.event.pull_request.base.ref || 'origin/main' }}
3.3. Matrix-Based Testing
The detect-changes.yml workflow outputs a list of changed modules, which is then transformed into a GitHub Actions matrix for parallel testing. This enables efficient, scalable test execution where each module runs independently:
Dynamic matrix generation from changed modules:
# .github/workflows/nf-test.yml
prepare-modules:
name: Prepare module matrix
needs: detect-changes
if: needs.detect-changes.outputs.modules != ''
runs-on: ubuntu-latest
outputs:
matrix: ${{ steps.set-matrix.outputs.matrix }}
steps:
- name: Set matrix
id: set-matrix
run: |
MODULES="${{ needs.detect-changes.outputs.modules }}"
# Convert space-separated string to JSON array
MATRIX=$(echo $MODULES | jq -cR 'split(" ") | map(select(length > 0))')
echo "matrix=$MATRIX" >> $GITHUB_OUTPUT
# Use the matrix for parallel jobs
nf-test-modules:
needs: [detect-changes, prepare-modules]
strategy:
matrix:
module: ${{ fromJson(needs.prepare-modules.outputs.matrix) }}
steps:
- name: Run nf-test with Docker
run: nf-test test modules/gianglabs/${{ matrix.module }}/ --profile docker
3.4. Practical Application: Reorganizing an Existing Pipeline
The pragmatic approach shines when migrating modules from an existing pipeline. As an example, the nf-germline-short-read-variant-calling pipeline was reorganized to extract reusable modules and subworkflows into gianglabs/nf-modules. This enables other pipelines to install these components directly:
# Install a specific module from gianglabs/nf-modules
nf-core modules --git-remote https://github.com/gianglabs/nf-modules.git install bcftools/index
# Install a reusable subworkflow
nf-core subworkflows --git-remote https://github.com/gianglabs/nf-modules.git install alignment
Repository structure after reorganization:
# Reorganized modules directory (v1.0.0)
modules/
├── gianglabs/ # Optimized for production use
│ ├── bcftools
│ ├── bedtools
│ ├── bwamem2
│ ├── fastp
│ ├── gatk
│ ├── gatkspark
│ ├── sambamba
│ ├── samtools
│ ├── snpeff
│ └── vep
└── local/ # Pipeline-specific modules
├── cnvnator
├── deepvariant
├── delly
├── freebayes
├── gatk
├── manta
├── smoove
├── survivor
└── tiddit
# Reusable subworkflows
subworkflows/
├── gianglabs/ # Domain-specific composition
│ ├── alignment
│ ├── alignment_preprocessing
│ ├── variant_alignment_quality_control
│ └── variant_annotation
└── local/ # Pipeline-specific logic
└── variant_calling
This separation enables code reuse across pipelines while maintaining pipeline-specific customizations in the local namespace.
References
- nf-core/modules — Official shared module repository
- gianglabs/nf-modules — Example custom modules
- nf-core/tools — Module management CLI tool
- nf-test Documentation — Testing framework
- Pixi Documentation — Reproducible environment management
- GATK Documentation — Genome Analysis Toolkit
- GitHub Actions Reusable Workflows — Workflow composition
Summary: The gianglabs/nf-modules repository demonstrates how domain-specific module libraries can balance the flexibility of nf-core with the practical constraints of production genomics workflows. By embracing pragmatic testing strategies, multi-language linting automation, and clear separation between reusable and pipeline-specific code, organizations can build efficient, maintainable, and scalable variant-calling pipelines.