GIAB Pilot Study: Establishing High-Confidence Genomic Benchmarks

The Genome in a Bottle (GIAB) Consortium has become the gold standard for variant calling benchmarking in clinical and research genomics. While many bioinformaticians routinely use GIAB reference materials to validate pipelines and assess variant caller performance, fewer understand the rigorous methodological framework underlying these benchmarks. This review examines the foundational 2014 pilot study by Zook et al., which established the technical infrastructure and integration strategies that continue to guide reference material development today.
Note on Figures: The original publication is not open-access. Figures presented here are derived from alternative public materials and community resources for educational purposes.
1. Motivation and Objectives
Core Principles of the GIAB Pilot Study:
- Utilize an immortalized cell line (NA12878) to ensure reproducible, unlimited DNA material across laboratories and time points
- Sequence the same genome using orthogonal technologies, diverse library preparation protocols, and independent analysis pipelines
- Define high-confidence variants through multi-platform consensus, requiring support from multiple independent technologies
- Systematically classify and exclude regions with conflicting calls or insufficient evidence
- Apply rigorous arbitration and manual curation to resolve discordances in challenging genomic regions
The clinical deployment of whole genome sequencing demands robust quality control frameworks capable of accurately characterizing genotypes at scale—spanning millions to billions of genomic positions. However, substantial inter-method discordance among contemporary sequencing technologies and bioinformatics pipelines posed a critical barrier to clinical translation. The field lacked a consensus reference standard for systematic performance assessment.
The seminal 2014 study by Zook et al. addressed this gap by establishing the first comprehensive, high-confidence benchmark for NA12878—a well-characterized HapMap/1000 Genomes CEU female sample subsequently adopted as a NIST reference material. The work introduced a principled integration methodology designed to minimize platform-specific and algorithmic biases through multi-technology arbitration, establishing a framework that continues to inform reference material development.
2. Methodological Framework
Selection of NA12878 as Reference Material:
- Cell type: EBV-transformed B-lymphocyte cell line enabling straightforward immortalization and robust culture scalability
- Biological stability: Immortalized cells provide consistent, renewable DNA source eliminating batch-to-batch variability
- Reproducibility: Enables repeated sequencing experiments with identical genomic input, facilitating cross-platform and cross-laboratory comparisons
To construct an unbiased, high-confidence variant call set, the study developed a sophisticated integration and arbitration framework leveraging orthogonal sequencing technologies and independent bioinformatics methodologies. This multi-platform consensus approach systematically identifies and mitigates technology-specific systematic errors and algorithmic biases.
2.1 Multi-Platform Data Integration Strategy
Historical Context: This pilot study was published in 2014. While some technologies (e.g., 454, SOLiD) are now deprecated, the integration principles remain highly relevant for modern benchmarking efforts incorporating contemporary platforms such as PacBio HiFi and Oxford Nanopore.
The integration strategy encompassed 14 independent datasets representing diverse sequencing chemistries and bioinformatics approaches:
- 5 sequencing platforms: Illumina (multiple instrument generations), Complete Genomics, Roche 454, Applied Biosystems SOLiD, and Ion Torrent
- 7 read alignment algorithms: BWA, BWA-MEM, Novoalign, SSAHA2, LifeScope, CGTools, and CASAVA
- 3 variant calling methods: GATK UnifiedGenotyper, GATK HaplotypeCaller, and Cortex (de novo assembly-based)
- Coverage depth: 11 whole-genome datasets (16-190× coverage) and 3 exome datasets (30-80× coverage)
2.2 Integration and Arbitration Pipeline
The multi-stage arbitration workflow systematically resolved inter-dataset discordances:
-
Comprehensive variant discovery: Aggregate all positions exhibiting evidence for SNPs or indels across any input dataset, establishing a complete candidate variant universe
-
Forced genotyping at union sites: Generate genotype calls from each dataset at all candidate loci, ensuring systematic evaluation across technologies even at sites with marginal evidence
-
Machine learning-based bias detection: Train GATK Variant Quality Score Recalibration (VQSR) using concordant calls to construct Gaussian mixture models identifying technology-specific systematic errors based on annotations including strand bias, mapping quality, allele balance, and positional biases
-
Sequential arbitration of discordances: Apply hierarchical filtering to resolve conflicting genotype calls, prioritizing exclusion of datasets exhibiting:
- Systematic sequencing errors (SSEs) characteristic of platform-specific artifacts
- Local alignment ambiguities in repetitive or low-complexity sequences
- Reduced mapping quality indicative of paralogous mapping
- Atypical allele balance suggesting technical artifacts or somatic mosaicism
-
Definition of uncertain regions: Systematically exclude genomic intervals precluding confident genotype assignment:
- Sites with persistent inter-dataset discordance after arbitration
- Simple repeats incompletely spanned by available read lengths
- Known segmental duplications and tandem repeat expansions
- Sequences paralogous to GRCh37 decoy contigs
- Reported structural variant breakpoints and complex rearrangements
3. Results
The final integrated call set achieved 87.6% coverage of non-N bases across chromosomes 1-22 and X, comprising:
- 2,484,884,293 high-confidence homozygous reference positions
- 3,137,725 SNP calls
- 201,629 indel calls (predominantly <40 bp)
Applying conservative structural variant exclusion reduced callable regions to 77% of the reference genome, retaining 2,741,014 SNPs and 174,718 indels. This conservative approach prioritizes specificity over sensitivity, ensuring benchmark utility for precision-focused applications.
4. Orthogonal Validation
The integrated call set underwent rigorous validation against multiple independent technologies:
- Sanger sequencing (gold standard): 100% concordance across 551 SNPs and 79 indels (XPrize: 124 SNPs, 37 indels; GeT-RM: 427 SNPs, 42 indels)
- Fosmid clone sequencing: High concordance across ~5% of the genome, with manual curation resolving discordances primarily attributable to fosmid false negatives in complex variant contexts
- Microarray genotyping: Systematic comparison revealed significant microarray limitations, particularly in low-complexity regions where probe hybridization is compromised
- Independent variant calling (FreeBayes on WES): Manual inspection confirmed that WES-specific calls absent from the WGS benchmark represented false positives driven by exome capture biases and systematic mapping errors, further validating the robustness of whole-genome integration
Quality metrics support the superior accuracy of integrated calls. The transition/transversion (Ti/Tv) ratio for novel variants exceeded that of individual datasets, indicating reduced false positive rates. While Ti/Tv is an imperfect metric (novel variants may exhibit different mutational spectra), the elevated ratios provide corroborating evidence of improved specificity.
5. Technical Challenges and Limitations
5.1 Complex Variant Representation Ambiguity
A fundamental challenge in variant integration is representation non-uniqueness: indels and complex variants (clustered SNPs/indels within ~20 bp) admit multiple valid VCF representations. Different alignment algorithms produce divergent but equivalent representations of identical underlying variants, complicating cross-platform comparison.
To address this, the study employed VCF normalization strategies (vcflib vcfallelicprimitives for left-alignment and allelic decomposition) and standardized representation to GATK HaplotypeCaller format, which performs local de novo assembly to produce consistent representations. Manual inspection of discordant sites remained essential to distinguish genuine variant differences from representational artifacts.
 Figure: Illustration of representation ambiguity. A single biological variant can be encoded in multiple equivalent VCF formats depending on alignment algorithm and variant caller conventions.
Figure: Illustration of representation ambiguity. A single biological variant can be encoded in multiple equivalent VCF formats depending on alignment algorithm and variant caller conventions.
5.2 Platform-Specific Systematic Errors
Each sequencing technology exhibited characteristic error profiles in specific genomic contexts:
- Illumina: Strand-specific substitution errors adjacent to GGT motifs, attributable to known cluster phasing issues in earlier sequencing chemistries
- Coverage heterogeneity: Technology-dependent GC bias and capture efficiency variations produce platform-specific low-coverage regions
- Mapping ambiguity: Repetitive and paralogous sequences exhibit technology-dependent mapping difficulty, correlating with read length and paired-end insert size distributions
Example: Illumina data revealed strand-biased variant calls proximal to GGT trinucleotide motifs. These putative variants exhibited systematic allele balance distortions and directional strand bias in paired-end reads—hallmarks of systematic sequencing artifacts rather than genuine polymorphisms.
Critically, these calls were absent in orthogonal platforms (454, SOLiD, Complete Genomics), conclusively demonstrating platform-specific false positives. This exemplifies the power of multi-technology arbitration: technology-specific systematic errors are readily identified and filtered through orthogonal validation, while true variants receive multi-platform support.
This principle remains central to contemporary benchmark development, now incorporating long-read platforms (PacBio HiFi, ONT) with orthogonal error profiles to Illumina short reads.
5.3 Microarray Validation Limitations
While historically employed for sequencing validation, microarray genotyping platforms proved inadequate for comprehensive benchmark validation:
- Biased genomic sampling: Only 0.0117% of microarray probe sites interrogate low-complexity regions, compared to 0.7847% of integrated sequencing variants, creating systematic ascertainment bias
- Probe interference: Proximal indels and phased variants confound probe hybridization kinetics, producing erroneous genotype calls
- High false positive rate: Manual curation revealed that approximately 50% of microarray-specific variant calls represented clear homozygous reference positions across all sequencing datasets, indicating substantial microarray error rates
These findings underscore that microarray data, while useful for assessing common variants in mappable regions, cannot serve as a comprehensive truth set for whole-genome benchmarking. Sequencing-based validation with orthogonal technologies provides superior accuracy and genomic breadth.
5.4 Indel Calling Challenges
Indel detection exhibited substantially greater technical difficulty than SNP calling across all platforms:
- Homopolymer sensitivity: Indels within homopolymer tracts and low-complexity sequences remain inherently difficult to resolve accurately due to alignment ambiguity and polymerase slippage errors
- Reduced accuracy: All platforms demonstrated elevated false positive and false negative rates for indels compared to SNPs, necessitating more stringent filtering thresholds
- Complex arbitration requirements: Resolving discordant indel calls required sophisticated strategies incorporating local realignment, de novo assembly, and extensive manual curation
These challenges persist in modern variant calling, though long-read technologies (PacBio HiFi) have substantially improved indel accuracy in repetitive contexts.
5.5 Genomic Coverage Limitations
Despite the comprehensive multi-platform integration, approximately 23% of the reference genome was classified as uncertain or excluded from high-confidence regions:
- Structural variant breakpoints (~10% of genome): Regions harboring reported SVs in dbVar were conservatively excluded
- Segmental duplications: High-identity paralogous sequences preclude confident variant calling with short-read technologies
- Low-complexity regions: Homopolymers, tandem repeats, and simple sequence repeats exhibit systematic alignment and calling difficulties
- Low mappability: Regions with insufficient uniqueness for unambiguous short-read alignment
- Simple tandem repeats: STR loci require specialized genotyping approaches
Additionally, indel calls were predominantly limited to <40 bp in length, leaving larger insertions, deletions, and complex structural variants uncharacterized. Subsequent GIAB releases have progressively expanded coverage into difficult regions using long-read sequencing and specialized SV calling methods.
6. Impact and Continuing Relevance
The GIAB pilot study established foundational principles for genomic benchmark development that remain relevant a decade later. By systematically integrating 14 datasets spanning five sequencing platforms, seven alignment algorithms, and three variant calling strategies, the consortium produced high-confidence genotype calls for NA12878 demonstrating superior sensitivity, specificity, and reduced bias compared to any single-technology approach.
Key Methodological Contributions
- Multi-technology arbitration framework: Established principled methods for identifying and mitigating platform-specific systematic errors through orthogonal technology validation
- Comprehensive genome coverage: Provided high-confidence calls across 77-87% of the reference genome with validated SNP and indel genotypes
- Empirical demonstration of integration value: Definitively showed that multi-platform consensus substantially outperforms single-platform calling
- Systematic characterization of difficult regions: Categorized and documented genomic intervals where short-read technologies cannot reliably call variants
Community Impact
The benchmark resources, publicly distributed via genomeinabottle.org and NCBI FTP sites, have enabled:
- Pipeline validation: Standardized assessment of variant calling sensitivity and precision across the bioinformatics community
- Technology benchmarking: Objective comparison of sequencing platforms and library preparation protocols
- Clinical validation: FDA-recognized reference materials supporting diagnostic assay validation
- Algorithm development: Ground truth datasets facilitating machine learning and variant caller optimization
Evolution and Future Directions
The methodological framework established in this pilot study continues to guide GIAB benchmark refinement. Subsequent releases have progressively incorporated:
- Long-read sequencing (PacBio HiFi, Oxford Nanopore) enabling characterization of previously intractable regions
- Specialized structural variant calling methods expanding coverage beyond small indels
- Additional reference genomes representing diverse populations and genomic backgrounds
- Improved coverage of medically relevant genes and difficult genomic regions
This work demonstrated that well-characterized, technology-agnostic reference materials are essential infrastructure for clinical genomics, establishing principles that remain central to quality control in precision medicine.
7. Practical Tool: Multi-Platform Variant Inspection
While the original publication is not open-access, the principles can be applied in practice through systematic multi-platform variant review. To facilitate this, I developed a genomics toolkit leveraging igv-reports for interactive browser-based visualization of variant calls across multiple datasets.
This toolkit automates the workflow of subsetting BAM/VCF files for regions of interest and generating integrated IGV reports, enabling rapid manual curation of discordant calls—a critical step in benchmark development and variant validation.
Usage Example
# Clone the genomics toolkit
git clone git@github.com:gianglabs/gkit.git
cd gkit/igv-report
# Test with example data (1 BAM + 1 VCF)
make test
# For custom analyses: subset BAM/VCF files to regions of interest
bash scripts/remote_subset.sh \
<region_of_interest> \
<flanking_size> \
<input_data_path> \
<output_directory>
# Generate interactive IGV report
create_report <vcf_file> \
--genome <reference_genome> \
--flanking <flanking_size> \
--track-config ./output/tracks_config.json \
--output <output.html> \
--standalone \
--title "Multi-Platform Variant Review"
Recommended Workflow
- Identify discordant variants between your pipeline and GIAB benchmarks
- Extract regions using the subsetting script with appropriate flanking (recommend 500-1000 bp)
- Include multiple BAM tracks representing different platforms/pipelines for comparison
- Review systematically for platform-specific biases, mapping artifacts, and representation issues
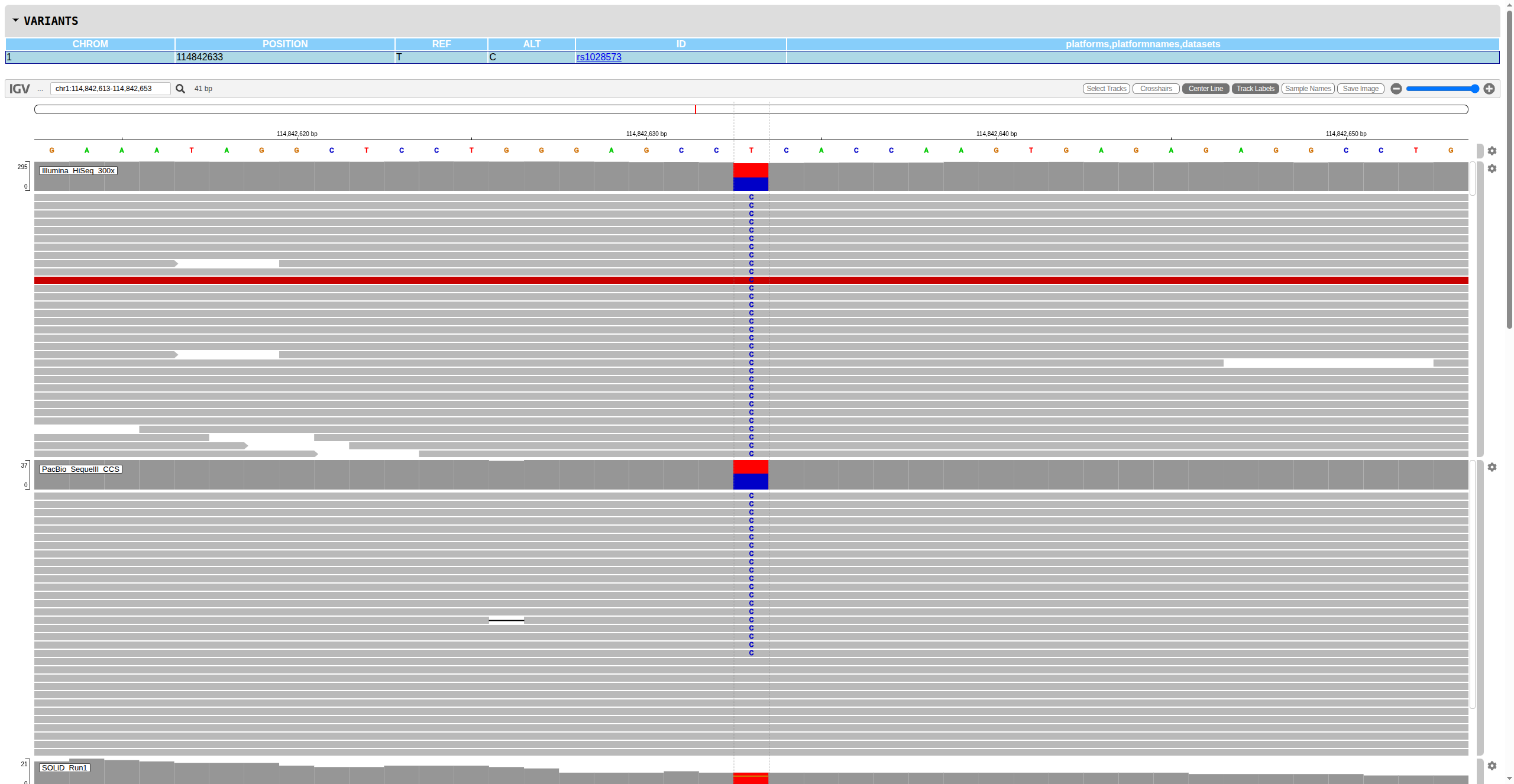 Figure: Example IGV report showing multi-platform alignment tracks for systematic variant review. Interactive browser-based visualization enables rapid assessment of variant support across technologies.
Figure: Example IGV report showing multi-platform alignment tracks for systematic variant review. Interactive browser-based visualization enables rapid assessment of variant support across technologies.
8. References
Primary Publication:
Zook, J. M., Chapman, B., Wang, J., Mittelman, D., Hofmann, O., Hide, W., & Salit, M. (2014). Integrating human sequence data sets provides a resource of benchmark SNP and indel genotype calls. Nature Biotechnology, 32(3), 246-251. https://doi.org/10.1038/nbt.2835
Resources:
- GIAB Consortium: https://www.genomeinabottle.org
- GIAB Data Repository: ftp://ftp-trace.ncbi.nlm.nih.gov/giab/ftp/
- IGV Reports: https://github.com/igvteam/igv-reports
- Genomics Toolkit (gkit): https://github.com/gianglabs/gkit