Variant Calling (Part 7): Variant Annotation with VEP and SnpSift: Integrating Functional Prediction and Variant Databases

After calling variants with high accuracy from the previous benchmark, the next step is variant annotation—understanding what each variant tells us about the sample. Variant annotation can be broadly categorized into two approaches: (1) using computational tools to predict the functional effect of variants, and (2) cross-referencing against databases of known variant effects.
- The environment setups and scripts are uploaded to nf-germline-short-read-variant-calling
- Cloning repository with tag 0.5.1 to reproduce this blog contents
This tutorial is for research only, for clinical application, it should be deeper in each tool configuration and curated database
1. Preparing Data and Reference Files for Annotation
1.1: Preparing Test Data
- When developing workflows, create test datasets that generate meaningful results
- A workflow may run successfully but produce no variants in the VCF file, making it difficult to validate the full functionality of the pipeline
- Use subset files smaller than 50MB that can be pushed to GitHub
Previously, I adapted the test data from nf-core/sarek; however, it did not generate any variants and was only useful for validating pipeline logic.
I previously used the HG002 30X coverage dataset for benchmarking. To create a subset for testing, I used the script below and placed it in the assets folder:
# raw fastq
samtools view -b HG002_merged.bam chr22:17000000-20000000 > subset.bam
samtools sort -n -o subset_name.bam subset.bam
samtools fastq \
-1 HG002_subset_R1.fastq.gz \
-2 HG002_subset_R2.fastq.gz \
-0 /dev/null \
-s /dev/null \
-n subset_name.bam
# subset genome
# download only chr22
aws s3 ls --no-sign-request s3://ngi-igenomes/igenomes/Homo_sapiens/GATK/GRCh38/Sequence/Chromosomes/chr22.fasta .
# index
samtools faidx chr22.fasta
# get fasta dict
picard CreateSequenceDictionary \
R=chr22.fasta \
O=chr22.dict
1.2 Genome Configuration
The required FASTA files have been updated in the assets folder. Additionally, SnpEff and VEP cache settings are configured as below.
- The
sarek_testgenome has been renamed totestand replaced with the chr22 human genome configuration
params {
genomes = [
'test': [
fasta: 'assets/genome/chr22.fasta',
fasta_fai: 'assets/genome/chr22.fasta.fai',
dict: 'assets/genome/chr22.dict',
index_bwa2_reference: true,
bwa2_index: 'null',
dbsnp: 'https://raw.githubusercontent.com/nf-core/test-datasets/modules/data/genomics/homo_sapiens/genome/vcf/dbsnp_146.hg38.vcf.gz',
dbsnp_tbi: 'https://raw.githubusercontent.com/nf-core/test-datasets/modules/data/genomics/homo_sapiens/genome/vcf/dbsnp_146.hg38.vcf.gz.tbi',
known_indels: 'https://raw.githubusercontent.com/nf-core/test-datasets/modules/data/genomics/homo_sapiens/genome/vcf/mills_and_1000G.indels.vcf.gz',
known_indels_tbi: 'https://raw.githubusercontent.com/nf-core/test-datasets/modules/data/genomics/homo_sapiens/genome/vcf/mills_and_1000G.indels.vcf.gz.tbi',
snpeff_cache: "s3://annotation-cache/snpeff_cache/GRCh38.99",
vep_cache_version : '115',
vep_genome : 'GRCh38',
vep_species : 'homo_sapiens',
vep_cache: "s3://annotation-cache/vep_cache/115_GRCh38/homo_sapiens/115_GRCh38/22"
],
'GRCh38': [
fasta: "s3://ngi-igenomes/igenomes/Homo_sapiens/GATK/GRCh38/Sequence/WholeGenomeFasta/Homo_sapiens_assembly38.fasta",
fasta_fai: "s3://ngi-igenomes/igenomes/Homo_sapiens/GATK/GRCh38/Sequence/WholeGenomeFasta/Homo_sapiens_assembly38.fasta.fai",
dict: "s3://ngi-igenomes/igenomes/Homo_sapiens/GATK/GRCh38/Sequence/WholeGenomeFasta/Homo_sapiens_assembly38.dict",
index_bwa2_reference: false,
bwa2_index: "s3://ngi-igenomes/igenomes/Homo_sapiens/GATK/GRCh38/Sequence/BWAmem2Index/",
dbsnp: "s3://ngi-igenomes/igenomes/Homo_sapiens/GATK/GRCh38/Annotation/GATKBundle/dbsnp_146.hg38.vcf.gz",
dbsnp_tbi: "s3://ngi-igenomes/igenomes/Homo_sapiens/GATK/GRCh38/Annotation/GATKBundle/dbsnp_146.hg38.vcf.gz.tbi",
known_indels: "s3://ngi-igenomes/igenomes/Homo_sapiens/GATK/GRCh38/Annotation/GATKBundle/Mills_and_1000G_gold_standard.indels.hg38.vcf.gz",
known_indels_tbi: "s3://ngi-igenomes/igenomes/Homo_sapiens/GATK/GRCh38/Annotation/GATKBundle/Mills_and_1000G_gold_standard.indels.hg38.vcf.gz.tbi",
known_indels_beta: "s3://ngi-igenomes/igenomes/Homo_sapiens/GATK/GRCh38/Annotation/GATKBundle/beta/Homo_sapiens_assembly38.known_indels.vcf.gz",
known_indels_beta_tbi: "s3://ngi-igenomes/igenomes/Homo_sapiens/GATK/GRCh38/Annotation/GATKBundle/beta/Homo_sapiens_assembly38.known_indels.vcf.gz.tbi",
snpeff_cache: "s3://annotation-cache/snpeff_cache/GRCh38.99",
vep_cache_version : '115',
vep_genome : 'GRCh38',
vep_species : 'homo_sapiens',
vep_cache: "s3://annotation-cache/vep_cache/115_GRCh38"
]
]
}
Except for the vep_cache parameter, which points to a public S3 bucket, the remaining VEP parameters are values that the annotation tools use later. For VEP annotation, the cache folder must be well-organized with subfolders for the cache version and genome.
You can list the available subfolders as shown below:
# list files in this folder (S3 prefix)
aws s3 ls --no-sign-request s3://annotation-cache/vep_cache/115_GRCh38/homo_sapiens/115_GRCh38
# PRE 115_GRCh38/
- Instead of downloading the entire database, specifying only chr22 for annotation is sufficient for testing (~300MB)
- The module includes bash script logic to handle the subfolder structure requirement
- Cache Bucket Access: Public S3 buckets can be accessed with
--no-sign-requestflag (no credentials needed) - Database Modes: VEP supports two modes:
- Cache mode (default): Pre-downloaded local cache files provide faster annotation without internet access, ideal for offline pipelines
- SQL mode (
--database): Requires live database connection (slower but always up-to-date), useful for single-run jobs where cache download overhead isn't justified
- Both modes can be combined with
--pick_allele_geneto select a single transcript per variant (more clinically relevant)
2. Modules and Subworkflows
2.1 Bcftools Normalize Variant
Called variants need to be normalized to address common issues:
| Issue | Example | Impact on Annotation |
|---|---|---|
| Not left-aligned | chr1:1001 AT>A | HGVS coordinates may be incorrect |
| Non-minimal allele representation | chr1:2001 TTA>TA | Annotation tools may interpret the variant differently |
| Multiallelic site | chr1:3000 A>C,G | Annotation may mix effects across alleles |
| Database mismatch | Same variant represented differently | dbSNP or gnomAD lookup may fail |
The new BCFTOOLS_NORM module:
process BCFTOOLS_NORM {
tag "$meta.id"
label 'process_medium'
container 'community.wave.seqera.io/library/bcftools:1.21--4335bec1d7b44d11' // v1.21 (wave.seqera.io pre-built)
input:
tuple val(meta), path(vcf), path(tbi)
path(fasta)
output:
tuple val(meta), path("${meta.id}.vcf.gz"), emit: vcf
tuple val(meta), path("${meta.id}.vcf.gz.tbi"), emit: tbi
path "versions.yml" , emit: versions
script:
"""
bcftools norm \\
--fasta-ref ${fasta} \\
--output ${meta.id}.vcf.gz \\
--output-type z \\
--threads $task.cpus \\
${vcf}
tabix -p vcf ${meta.id}.vcf.gz
cat <<-END_VERSIONS > versions.yml
"${task.process}":
bcftools: \$(bcftools --version 2>&1 | head -n1 | sed 's/^.*bcftools //; s/ .*\$//')
END_VERSIONS
"""
}
2.2 SnpEff
SnpEff combines reference gene and transcript information to predict the functional effect of variants. It supports multiple databases; here, I used the Ensembl database:
process SNPEFF {
tag "${meta.id}"
label 'process_medium'
container 'quay.io/biocontainers/snpeff:5.4.0c--hdfd78af_0'
input:
tuple val(meta), path(vcf), path(vcf_tbi)
path(cache)
output:
tuple val(meta), path("*.ann.vcf"), emit: vcf
tuple val(meta), path("*.csv"), emit: report
tuple val(meta), path("*.html"), emit: summary_html
tuple val(meta), path("*.genes.txt"), emit: genes_txt
path "versions.yml", emit: versions
when:
task.ext.when == null || task.ext.when
script:
def avail_mem = 6144
if (!task.memory) {
log.info('[snpEff] Available memory not known - defaulting to 6GB. Specify process memory requirements to change this.')
}
else {
avail_mem = (task.memory.mega * 0.8).intValue()
}
def prefix = task.ext.prefix ?: "${meta.id}"
"""
snpEff \\
-Xmx${avail_mem}M \\
${cache} \\
-nodownload -canon -v \\
-csvStats ${prefix}.csv \\
-dataDir \${PWD}/${cache} \\
${vcf} \\
> ${prefix}.ann.vcf
cat <<-END_VERSIONS > versions.yml
"${task.process}":
snpeff: \$(snpEff -version 2>&1 | grep -oP 'version [0-9.]+' | sed 's/version //' || echo "5.1")
END_VERSIONS
"""
}
2.3 VEP
As described above, the VEP cache directory must be well-organized with subfolders for the cache version and genome. This can be accomplished in three steps:
- Create a temporary cache folder with the cache version and genome structure
- Find the
all_vars.gzfile in the cache input path and copy its subfolders to the temporary cache folder - Use this temporary cache folder for the VEP command-line argument
process VEP {
tag "${meta.id}"
label 'process_medium'
container 'community.wave.seqera.io/library/ensembl-vep_perl-math-cdf:1e13f65f931a6954'
input:
tuple val(meta), path(vcf)
path cache
val cache_version
val genome
val species
path fasta
output:
tuple val(meta), path("*.vcf.gz"), emit: vcf, optional: true
tuple val(meta), path("*.vcf.gz.tbi"), emit: tbi, optional: true
tuple val(meta), path("*.tab.gz"), emit: tab, optional: true
tuple val(meta), path("*.json.gz"), emit: json, optional: true
tuple val(meta), val("${task.process}"), val('ensemblvep'), path("*.html"), topic: multiqc_files, emit: report, optional: true
tuple val("${task.process}"), val('ensemblvep'), eval("vep --help | sed -n '/ensembl-vep/s/.*: //p'"), topic: versions, emit: versions_ensemblvep
path "versions.yml", emit: versions
script:
def temp_vep_cache = "temp_vep_cache/${species}/${cache_version}_${genome}"
"""
mkdir -p ${temp_vep_cache}
cache_dirname=\$(find -L $cache -name all_vars.gz -print -quit | xargs dirname)
mv \$cache_dirname ${temp_vep_cache}
vep \\
-i ${vcf} \\
-o ${meta.id}.vcf.gz \\
--stats_file ${meta.id}.html --vcf --everything --filter_common --per_gene --total_length --offline --format vcf \\
--compress_output bgzip \\
--assembly ${genome} \\
--species ${species} \\
--cache \\
--fasta ${fasta} \\
--cache_version ${cache_version} \\
--dir_cache \$PWD/temp_vep_cache \\
--fork ${task.cpus}
tabix -p vcf ${meta.id}.vcf.gz
cat <<-END_VERSIONS > versions.yml
"${task.process}":
ensembl-vep: \$(vep --version 2>&1 | grep -oP 'ensembl-vep [0-9.]+' | sed 's/ensembl-vep //' || echo "104.3")
END_VERSIONS
"""
}
2.4 New Annotation Subworkflow
The subworkflow is created as shown below and accepts input from the variant calling step:
#!/usr/bin/env nextflow
nextflow.enable.dsl = 2
/*
========================================================================================
ANNOTATION SUBWORKFLOW
========================================================================================
*/
// Include modules
include { BCFTOOLS_NORM } from '../../../modules/local/bcftools/norm/main'
include { SNPEFF } from '../../../modules/local/snpeff/main'
include { VEP } from '../../../modules/local/vep/main'
workflow VARIANT_ANNOTATION {
take:
vcf // channel: [ val(meta), path(vcf) ]
vcf_tbi // channel: [ val(meta), path(tbi) ]
snpeff_cache // value
vep_cache // value
vep_cache_version // value
vep_genome // value
vep_species // value
fasta // path to reference fasta
main:
ch_versions = channel.empty()
ch_snpeff_cache_db = channel.fromPath(snpeff_cache, checkIfExists: true).collect()
ch_vep_cache_db = channel.fromPath(vep_cache, checkIfExists: true).collect()
BCFTOOLS_NORM(
vcf.join(vcf_tbi),
fasta
)
ch_versions = ch_versions.mix(BCFTOOLS_NORM.out.versions)
SNPEFF(
BCFTOOLS_NORM.out.vcf.join(BCFTOOLS_NORM.out.tbi),
ch_snpeff_cache_db
)
ch_versions = ch_versions.mix(SNPEFF.out.versions)
VEP(
SNPEFF.out.vcf,
ch_vep_cache_db,
vep_cache_version,
vep_genome,
vep_species,
fasta
)
ch_versions = ch_versions.mix(VEP.out.versions)
emit:
annotated_vcf = VEP.out.vcf // channel: [ val(meta), path(annotated_vcf), path(annotated_tbi) ]
versions = ch_versions
}
The main.nf workflow now includes the new VARIANT_ANNOTATION step:
...
VARIANT_ANNOTATION(
VARIANT_CALLING.out.vcf,
VARIANT_CALLING.out.vcf_tbi,
params.snpeff_cache,
params.vep_cache,
params.vep_cache_version,
params.vep_genome,
params.vep_species,
ref_fasta
)
...
3. Run the Updated Workflow and Evaluate Variant Annotation
3.1 Run the Workflow With Test Profile
make test-e2e
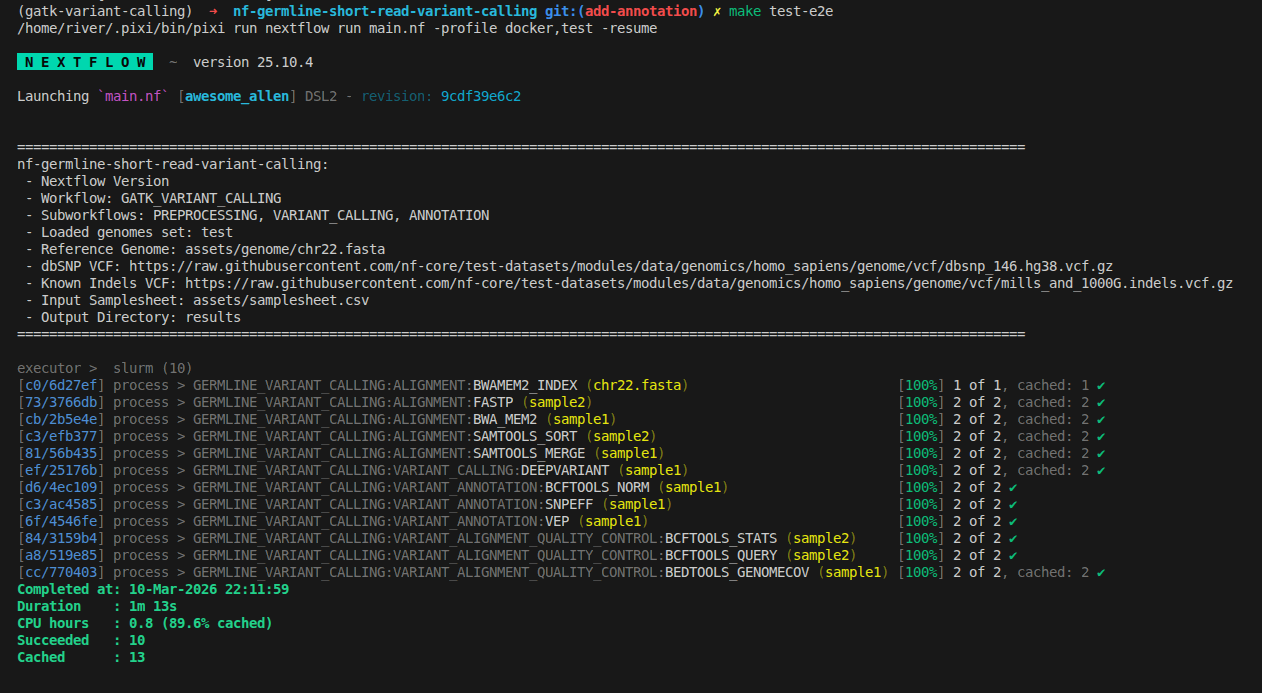
3.2 Validate the Variant Annotation
3.2.1 Called Variants
To view the VCF header with descriptions for columns in the VCF file, use: bcftools view --header-only <vcf file>
The variants are called by DeepVariant:
zcat results/germline_variant_calling:variant_calling:deepvariant/sample1.vcf.gz|tail -n5
Example output:
chr22 21326676 . A G 11.2 PASS . GT:GQ:DP:AD:VAF:PL 1/1:11:6:0,6:1:10,19,0
chr22 32146109 . T A 0 RefCall . GT:GQ:DP:AD:VAF:PL 0/0:20:7:0,6:0.857143:0,23,22
chr22 32146116 . A T 0.1 RefCall . GT:GQ:DP:AD:VAF:PL ./.:16:7:0,7:1:0,20,18
chr22 32146120 . A G 0.1 RefCall . GT:GQ:DP:AD:VAF:PL ./.:17:7:0,7:1:0,21,19
chr22 32146129 . T A 0.1 RefCall . GT:GQ:DP:AD:VAF:PL ./.:19:7:0,7:1:0,22,21
3.2.2 SnpEff Annotation
Get the last 5 lines:
tail -n5 results/germline_variant_calling:variant_annotation:snpeff/sample1.ann.vcf
The additional columns show HGVS notation and indicate where the variant is located relative to nearby genes:
chr22 21326676 . A G 11.2 PASS ANN=G|intergenic_region|MODIFIER|FAM230H-PPP1R26P5|ENSG00000206142-ENSG00000232771|intergenic_region|ENSG00000206142-ENSG00000232771|||n.21326676A>G|||||| GT:GQ:DP:AD:VAF:PL 1/1:11:6:0,6:1:10,19,0
chr22 32146109 . T A 0.0 RefCall ANN=A|downstream_gene_variant|MODIFIER|C22orf42|ENSG00000205856|transcript|ENST00000382097.4|protein_coding||c.*3431A>T|||||2897|,A|downstream_gene_variant|MODIFIER|Z83839.2|ENSG00000239674|transcript|ENST00000436294.1|pseudogene||n.*2837T>A|||||2837|,A|intergenic_region|MODIFIER|Z83839.2-C22orf42|ENSG00000239674-ENSG00000205856|intergenic_region|ENSG00000239674-ENSG00000205856|||n.32146109T>A|||||| GT:GQ:DP:AD:VAF:PL 0/0:20:7:0,6:0.857143:0,23,22
chr22 32146116 . A T 0.1 RefCall ANN=T|downstream_gene_variant|MODIFIER|C22orf42|ENSG00000205856|transcript|ENST00000382097.4|protein_coding||c.*3424T>A|||||2890|,T|downstream_gene_variant|MODIFIER|Z83839.2|ENSG00000239674|transcript|ENST00000436294.1|pseudogene||n.*2844A>T|||||2844|,T|intergenic_region|MODIFIER|Z83839.2-C22orf42|ENSG00000239674-ENSG00000205856|intergenic_region|ENSG00000239674-ENSG00000205856|||n.32146116A>T||||||;CSQ=T|downstream_gene_variant|MODIFIER|AP1B1P1|ENSG00000290475|Transcript|ENST00000716525|lncRNA|||||||||||2721|1||SNV|EntrezGene|HGNC:297|YES||||||||||||||||||||||||||||||||||||||||||||||||||||||,T|downstream_gene_variant|MODIFIER|C22orf42|ENSG00000205856|Transcript|ENST00000382097|protein_coding|||||||||||2890|-1||SNV|HGNC|HGNC:27160|YES|MANE_Select|NM_001010859.3||1|P1|CCDS33639.1|ENSP00000371529|Q6IC83.120||UPI00003765B0|||||||||||||||||||||||||||||||||||||||||||| GT:GQ:DP:AD:VAF:PL ./.:16:7:0,7:1:0,20,18
chr22 32146120 . A G 0.1 RefCall ANN=G|downstream_gene_variant|MODIFIER|C22orf42|ENSG00000205856|transcript|ENST00000382097.4|protein_coding||c.*3420T>C|||||2886|,G|downstream_gene_variant|MODIFIER|Z83839.2|ENSG00000239674|transcript|ENST00000436294.1|pseudogene||n.*2848A>G|||||2848|,G|intergenic_region|MODIFIER|Z83839.2-C22orf42|ENSG00000239674-ENSG00000205856|intergenic_region|ENSG00000239674-ENSG00000205856|||n.32146120A>G||||||;CSQ=G|downstream_gene_variant|MODIFIER|AP1B1P1|ENSG00000290475|Transcript|ENST00000716525|lncRNA|||||||||||2725|1||SNV|EntrezGene|HGNC:297|YES||||||||||||||||||||||||||||||||||||||||||||||||||||||,G|downstream_gene_variant|MODIFIER|C22orf42|ENSG00000205856|Transcript|ENST00000382097|protein_coding|||||||||||2886|-1||SNV|HGNC|HGNC:27160|YES|MANE_Select|NM_001010859.3||1|P1|CCDS33639.1|ENSP00000371529|Q6IC83.120||UPI00003765B0|||||||||||||||||||||||||||||||||||||||||||| GT:GQ:DP:AD:VAF:PL ./.:17:7:0,7:1:0,21,19
chr22 32146129 . T A 0.1 RefCall ANN=A|downstream_gene_variant|MODIFIER|C22orf42|ENSG00000205856|transcript|ENST00000382097.4|protein_coding||c.*3411A>T|||||2877|,A|downstream_gene_variant|MODIFIER|Z83839.2|ENSG00000239674|transcript|ENST00000436294.1|pseudogene||n.*2857T>A|||||2857|,A|intergenic_region|MODIFIER|Z83839.2-C22orf42|ENSG00000239674-ENSG00000205856|intergenic_region|ENSG00000239674-ENSG00000205856|||n.32146129T>A||||||;CSQ=A|downstream_gene_variant|MODIFIER|AP1B1P1|ENSG00000290475|Transcript|ENST00000716525|lncRNA|||||||||||2734|1||SNV|EntrezGene|HGNC:297|YES||||||||||||||||||||||||||||||||||||||||||||||||||||||,A|downstream_gene_variant|MODIFIER|C22orf42|ENSG00000205856|Transcript|ENST00000382097|protein_coding|||||||||||2877|-1||SNV|HGNC|HGNC:27160|YES|MANE_Select|NM_001010859.3||1|P1|CCDS33639.1|ENSP00000371529|Q6IC83.120||UPI00003765B0|||||||||||||||||||||||||||||||||||||||||||| GT:GQ:DP:AD:VAF:PL ./.:19:7:0,7:1:0,22,21
3.2.3 VEP Annotation
View the last 5 rows:
zcat results/germline_variant_calling:variant_annotation:vep/sample1.vcf.gz|tail -n5
The result shows the additional annotated columns in the last 5 rows:
chr22 21326676 . A G 11.2 PASS ANN=G|intergenic_region|MODIFIER|FAM230H-PPP1R26P5|ENSG00000206142-ENSG00000232771|intergenic_region|ENSG00000206142-ENSG00000232771|||n.21326676A>G||||||;CSQ=G|upstream_gene_variant|MODIFIER|FAM230H|ENSG00000206142|Transcript|ENST00000447720|lncRNA|||||||||||1634|-1||SNV|HGNC|HGNC:53977|YES||||5||||||||||||||||||||||||||||||||||||||||||||||||||,G|upstream_gene_variant|MODIFIER||ENSG00000303017|Transcript|ENST00000791156|lncRNA|||||||||||50|1||SNV|||YES|||||||||||||||||||||||||||||||||||||||||||||||||||||| GT:GQ:DP:AD:VAF:PL 1/1:11:6:0,6:1:10,19,0
chr22 32146109 . T A 0.0 RefCall ANN=A|downstream_gene_variant|MODIFIER|C22orf42|ENSG00000205856|transcript|ENST00000382097.4|protein_coding||c.*3431A>T|||||2897|,A|downstream_gene_variant|MODIFIER|Z83839.2|ENSG00000239674|transcript|ENST00000436294.1|pseudogene||n.*2837T>A|||||2837|,A|intergenic_region|MODIFIER|Z83839.2-C22orf42|ENSG00000239674-ENSG00000205856|intergenic_region|ENSG00000239674-ENSG00000205856|||n.32146109T>A||||||;CSQ=A|downstream_gene_variant|MODIFIER|AP1B1P1|ENSG00000290475|Transcript|ENST00000716525|lncRNA|||||||||||2714|1||SNV|EntrezGene|HGNC:297|YES||||||||||||||||||||||||||||||||||||||||||||||||||||||,A|downstream_gene_variant|MODIFIER|C22orf42|ENSG00000205856|Transcript|ENST00000382097|protein_coding|||||||||||2897|-1||SNV|HGNC|HGNC:27160|YES|MANE_Select|NM_001010859.3||1|P1|CCDS33639.1|ENSP00000371529|Q6IC83.120||UPI00003765B0|||||||||||||||||||||||||||||||||||||||||||| GT:GQ:DP:AD:VAF:PL 0/0:20:7:0,6:0.857143:0,23,22
chr22 32146116 . A T 0.1 RefCall ANN=T|downstream_gene_variant|MODIFIER|C22orf42|ENSG00000205856|transcript|ENST00000382097.4|protein_coding||c.*3424T>A|||||2890|,T|downstream_gene_variant|MODIFIER|Z83839.2|ENSG00000239674|transcript|ENST00000436294.1|pseudogene||n.*2844A>T|||||2844|,T|intergenic_region|MODIFIER|Z83839.2-C22orf42|ENSG00000239674-ENSG00000205856|intergenic_region|ENSG00000239674-ENSG00000205856|||n.32146116A>T||||||;CSQ=T|downstream_gene_variant|MODIFIER|AP1B1P1|ENSG00000290475|Transcript|ENST00000716525|lncRNA|||||||||||2721|1||SNV|EntrezGene|HGNC:297|YES||||||||||||||||||||||||||||||||||||||||||||||||||||||,T|downstream_gene_variant|MODIFIER|C22orf42|ENSG00000205856|Transcript|ENST00000382097|protein_coding|||||||||||2890|-1||SNV|HGNC|HGNC:27160|YES|MANE_Select|NM_001010859.3||1|P1|CCDS33639.1|ENSP00000371529|Q6IC83.120||UPI00003765B0|||||||||||||||||||||||||||||||||||||||||||| GT:GQ:DP:AD:VAF:PL ./.:16:7:0,7:1:0,20,18
chr22 32146120 . A G 0.1 RefCall ANN=G|downstream_gene_variant|MODIFIER|C22orf42|ENSG00000205856|transcript|ENST00000382097.4|protein_coding||c.*3420T>C|||||2886|,G|downstream_gene_variant|MODIFIER|Z83839.2|ENSG00000239674|transcript|ENST00000436294.1|pseudogene||n.*2848A>G|||||2848|,G|intergenic_region|MODIFIER|Z83839.2-C22orf42|ENSG00000239674-ENSG00000205856|intergenic_region|ENSG00000239674-ENSG00000205856|||n.32146120A>G||||||;CSQ=G|downstream_gene_variant|MODIFIER|AP1B1P1|ENSG00000290475|Transcript|ENST00000716525|lncRNA|||||||||||2725|1||SNV|EntrezGene|HGNC:297|YES||||||||||||||||||||||||||||||||||||||||||||||||||||||,G|downstream_gene_variant|MODIFIER|C22orf42|ENSG00000205856|Transcript|ENST00000382097|protein_coding|||||||||||2886|-1||SNV|HGNC|HGNC:27160|YES|MANE_Select|NM_001010859.3||1|P1|CCDS33639.1|ENSP00000371529|Q6IC83.120||UPI00003765B0|||||||||||||||||||||||||||||||||||||||||||| GT:GQ:DP:AD:VAF:PL ./.:17:7:0,7:1:0,21,19
chr22 32146129 . T A 0.1 RefCall ANN=A|downstream_gene_variant|MODIFIER|C22orf42|ENSG00000205856|transcript|ENST00000382097.4|protein_coding||c.*3411A>T|||||2877|,A|downstream_gene_variant|MODIFIER|Z83839.2|ENSG00000239674|transcript|ENST00000436294.1|pseudogene||n.*2857T>A|||||2857|,A|intergenic_region|MODIFIER|Z83839.2-C22orf42|ENSG00000239674-ENSG00000205856|intergenic_region|ENSG00000239674-ENSG00000205856|||n.32146129T>A||||||;CSQ=A|downstream_gene_variant|MODIFIER|AP1B1P1|ENSG00000290475|Transcript|ENST00000716525|lncRNA|||||||||||2734|1||SNV|EntrezGene|HGNC:297|YES||||||||||||||||||||||||||||||||||||||||||||||||||||||,A|downstream_gene_variant|MODIFIER|C22orf42|ENSG00000205856|Transcript|ENST00000382097|protein_coding|||||||||||2877|-1||SNV|HGNC|HGNC:27160|YES|MANE_Select|NM_001010859.3||1|P1|CCDS33639.1|ENSP00000371529|Q6IC83.120||UPI00003765B0|||||||||||||||||||||||||||||||||||||||||||| GT:GQ:DP:AD:VAF:PL ./.:19:7:0,7:1:0,22,21
The variant annotation is generated by combining results from SnpEff (ANN field) and Ensembl VEP (CSQ field). Each annotation describes the predicted functional consequence of the variant relative to known genes and transcripts in the GRCh38 reference genome. Because a variant may overlap multiple transcripts or nearby genes, multiple annotations can appear for a single variant.
| Field | Example value | Source | Description |
|---|---|---|---|
| Allele | A | ANN / CSQ | Alternate allele being annotated |
| Consequence | downstream_gene_variant | ANN / CSQ | Predicted effect relative to gene structure |
| Impact | MODIFIER | ANN / CSQ | Impact category (HIGH, MODERATE, LOW, MODIFIER) |
| Gene symbol | C22orf42 | ANN / CSQ | Gene name |
| Gene ID | ENSG00000205856 | ANN / CSQ | Ensembl gene identifier |
| Feature type | transcript | ANN / CSQ | Type of genomic feature |
| Transcript ID | ENST00000382097 | ANN / CSQ | Ensembl transcript identifier |
| Biotype | protein_coding | ANN / CSQ | Transcript functional class |
| Rank | - | ANN | Exon/intron rank within transcript |
| HGVS.c | c.*3411A>T | ANN | HGVS coding DNA notation |
| Distance | 2877 | ANN / CSQ | Distance to the nearest gene feature (bp) |
| Variant class | SNV | CSQ | Type of variant (single nucleotide variant) |
| Strand | -1 | CSQ | Gene strand orientation |
| Gene source | HGNC | CSQ | Gene symbol authority |
| HGNC ID | HGNC:27160 | CSQ | HGNC gene identifier |
| Canonical transcript | YES | CSQ | Transcript considered canonical by Ensembl |
| MANE Select | MANE_Select | CSQ | Matched RefSeq/Ensembl transcript |
| RefSeq ID | NM_001010859.3 | CSQ | Corresponding RefSeq transcript |
| CCDS | CCDS33639.1 | CSQ | Consensus CDS identifier |
| Protein ID | ENSP00000371529 | CSQ | Ensembl protein identifier |
| UniProt | Q6IC83 | CSQ | Protein identifier in UniProt |
| Population frequency | gnomAD fields | CSQ | Allele frequency in population datasets |
Recap
This blog presents a proof-of-concept variant annotation workflow using SnpEff and Ensembl VEP. While functional for research and discovery purposes, clinical implementation requires additional considerations:
Clinical Application & ACMG/AMP Guidelines
- ACMG/AMP Classification: Variants must be classified according to ACMG/AMP standards (benign, likely benign, uncertain significance, likely pathogenic, pathogenic)
- Evidence aggregation: Combine computational predictions, population frequencies, and literature evidence
- Quality control: Implement variant filtering based on quality scores, depth, and allele balance
- Validation: Independent validation through orthogonal methods (Sanger sequencing, PCR)
Database Integration
The annotation workflow can be expanded to integrate additional clinical databases:
- ClinVar: Clinically relevant variants with evidence levels and assertions
- gnomAD/ExAC: Population allele frequencies for benign variant filtering
- COSMIC: Cancer-associated somatic variants
- dbSNP/rs IDs: Known polymorphisms
- OMIM: Gene-phenotype associations
- UniProt/Protein databases: Functional protein annotations
- HGMD: Human Gene Mutation Database for inherited disease variants
- PharmGKB: Pharmacogenomic variants for drug response prediction
Future Enhancements
- Implement automated ACMG/AMP classification algorithms
- Add functional prediction scores (PolyPhen, SIFT, ConservationScores)
- Integrate pathway and network analysis
- Implement variant filtering pipelines for clinical reporting
- Add real-time database synchronization for latest evidence
- Develop multi-sample cohort analysis capabilities
This foundation enables expansion into comprehensive clinical genomics workflows while maintaining reproducibility and version control through containerization and infrastructure-as-code practices.
References
- SnpEff: https://pcingola.github.io/SnpEff
- VEP: https://ensembl.org/info/docs/tools/vep/index.html
- ACMG/AMP Guidelines: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4544753/
- ClinVar: https://www.ncbi.nlm.nih.gov/clinvar/
- gnomAD: https://gnomad.broadinstitute.org/