Variant Calling (Part 2): From Bash to Nextflow: GATK Best Practice With Nextflow

In Part 1, we built a complete 10-step GATK variant calling pipeline in bash—perfect for academic research and 1-10 samples. But what happens when you need to scale to 100+ samples? This is where Nextflow becomes essential.
📁 Repository: All code from this tutorial is organized in the nf-germline-short-read-variant-calling repository. The structure follows best practices with separate directories for bash (
bash-gatk) and Nextflow (nextflow-gatk) implementations.
This tutorial has been released under Github repository: https://github.com/nttg8100/nf-germline-short-read-variant-calling/tree/0.1.0
This blog simply shows you how the bash script can be transformed to nextflow, and how you can ensure the consistent results between the migration
1. Why Migrate from Bash to Nextflow?
1.1. The Bash Approach (Part 1)
Our bash pipeline works great for:
- Small-scale analysis: 1-10 samples
- Academic research: Quick prototypes and proof-of-concept
- Learning: Understanding GATK best practices step-by-step
- Local execution: Single machine, serial processing
Runtime for 1000 samples: ~50 hours (serial execution)
1.2. The Nextflow Approach (Part 2)
Nextflow enables:
- Large-scale production: 100+ samples in parallel
- HPC/Cloud execution: AWS Batch, Google Cloud, Slurm, SGE, etc.
- Automatic resume: Failed jobs restart from last checkpoint (
-resume) - Reproducibility: Containers, version control, audit trails
- Scalability: Processes run in parallel across compute nodes
Runtime for 1000 samples: ~2-3 hours (100x parallelization)
2. Run Bash Pipeline and Generate Baseline
Run the complete bash pipeline from Part 1:
# Clone and setup repository (first time only)
git clone git@github.com:nttg8100/nf-germline-short-read-variant-calling.git -b 0.1.0
cd nf-germline-short-read-variant-calling
# Install dependencies
pixi shell --manifest-path bash-gatk
# Download test data
bash scripts/download_data.sh
# Run pipeline
bash bash-gatk/gatk_pipeline.sh
3. Migrate to Nextflow
3.1 Design Patterns
Modern workflow management frameworks (like Nextflow, Snakemake, Cromwell) share several core design patterns:
3.1.1 Template-Based Scripting
Define a script template with placeholders for variables. The workflow engine fills in these placeholders with actual values at runtime.
Example:
fastqc {{fastq_r1}} {{fastq_r2}}
3.1.2 Containerization
Containers ensure reproducible environments and consistent results across different systems.
Example:
# Pull container image
singularity pull quay.io/biocontainers/fastqc:0.12.1--hdfd78af_0
# Run FastQC inside the container
singularity run fastqc_0.12.1--hdfd78af_0.sif fastqc {{fastq_r1}} {{fastq_r2}}
3.1.3 Scheduling and Resource Management
Schedulers optimize resource usage by running multiple tasks in parallel, respecting CPU and memory limits. They also support execution on clusters or cloud platforms, manage dependencies, and trigger downstream steps only when prerequisites are complete.
- Run multiple samples concurrently (e.g., 4 samples at a time with 2 CPUs and 4GB RAM each)
- Distribute tasks across nodes in a cluster or cloud
- Automatically handle step dependencies and execution order
3.2. Nextflow Project Overview
The final clean Nextflow project (already available in nextflow-gatk):
nextflow-gatk
├── main.nf
├── modules
│ ├── bwa_mem.nf
│ ├── fastqc.nf
│ ├── gatk_applybqsr.nf
│ ├── gatk_baserecalibrator.nf
│ ├── gatk_collectmetrics.nf
│ ├── gatk_genotypegvcfs.nf
│ ├── gatk_haplotypecaller.nf
│ ├── gatk_markduplicates.nf
│ ├── samtools_sort.nf
│ └── trim_galore.nf
├── nextflow.config
├── pixi.lock
├── pixi.toml
Right here, it is simply split each step into a module then import to the main.nf. With this structure, it allows better to reuse the module
3.2. First Step Workflow
Create main.nf
On a basic process, it needs:
- process pattern: process <<PROCESS_NAME>>: Define your process
- input: define channels to be the inputs, it can be file, string, number or combination of them which is defined as
tuple-similar to dictionary in python - output: define the channels output, it has regular expression that collects files on the output files
- script: define the code block template that help to ingest the variables from inputs, processes configuration that generate the real script to use later
#!/usr/bin/env nextflow
nextflow.enable.dsl=2 // enable DSL2
// Define the module as template
process FASTQC {
input:
tuple val(meta), path(reads) // input variable, example: [sample, [/path/read1.fq, /path/read2.fq ]]
output:
tuple val(meta), path("*.html") // output variable, example: [sample, [/path/read1.html, /path/read2.html ]]
script: // template that has been mentioned above
"""
# Quality Control with FastQC
# This generates HTML and ZIP files with quality metrics
fastqc $reads
"""
}
// You need to define the workflow to call to tool to run
workflow {
// Prepare the channel which later is allow the workflow to know how to get input, output to run
ch_input = Channel.of([ params.sample , [file(params.fastq_r1), file(params.fastq_r2)] ]) // For file, we simply add with file()
// ch_input.view() // print statement that you can view what it is, ex: [sample1, [/nf-core/test-datasets/modules/data/genomics/homo_sapiens/illumina/fastq/test_1.fastq.gz, /nf-core/test-datasets/modules/data/genomics/homo_sapiens/illumina/fastq/test_2.fastq.gz]]
// Run workflow
FASTQC (
ch_input
)
}
You can create folder
# env setup
mkdir -p temp
cd temp
pixi init
pixi add nextflow singularity
pixi shell
pixi add fastqc
# add the workflow, check folder structure
tree .
├── main.nf
├── pixi.lock
└── pixi.toml
# now you can run it
nextflow run main.nf \
--sample sample1 \
--fastq_r1 https://raw.githubusercontent.com/nf-core/test-datasets/modules/data/genomics/homo_sapiens/illumina/fastq/test_1.fastq.gz \
--fastq_r2 https://raw.githubusercontent.com/nf-core/test-datasets/modules/data/genomics/homo_sapiens/illumina/fastq/test_2.fastq.gz
# not only nextflow, but many tool will help you to resume your work, ignore re-run if it was ran before.
# with nextflow, it can be easy done by adding `-resume`
nextflow run main.nf -resume \
--sample sample1 \
--fastq_r1 https://raw.githubusercontent.com/nf-core/test-datasets/modules/data/genomics/homo_sapiens/illumina/fastq/test_1.fastq.gz \
--fastq_r2 https://raw.githubusercontent.com/nf-core/test-datasets/modules/data/genomics/homo_sapiens/illumina/fastq/test_2.fastq.gz
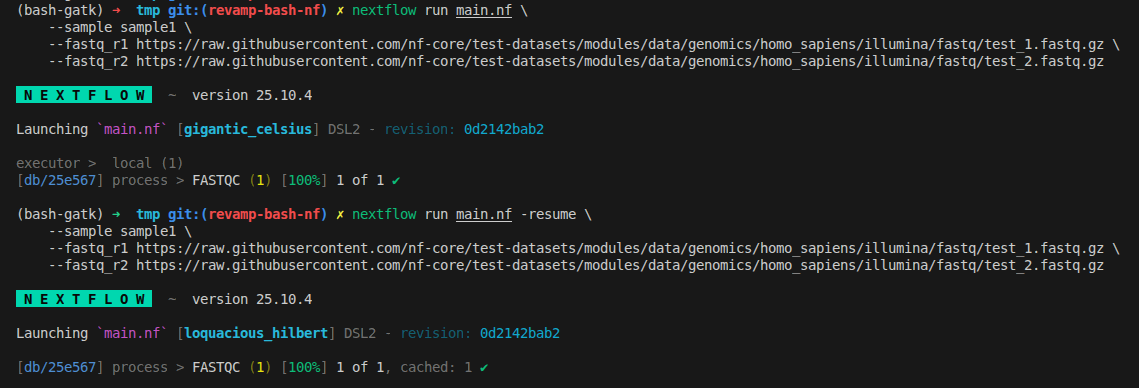
What we can see now is the automation from the nextflow
- It downloads files remotely
- It will create a script that automatically run your script with specific setup
- It will generate a folder called
workwhere it has subfolder that hashes for each tasks. Inside, without nextflow we can run independently
3.3 Deeper In Task Caching with Native Workflow
Now you can change working directory to the work folder to see its structure
As I mentioned above, what it does is just creating a work folder.
Here’s what each file in the Nextflow work directory means:
.command.begin: Marker file indicating when the task started..command.err: Captures standard error output (stderr) from the process—useful for debugging errors..command.log: Combined log of both stdout and stderr for the process execution..command.out: Captures standard output (stdout) from the process..command.run: Nextflow’s infrastructure wrapper script; handles environment setup, file staging, container execution, error handling, and runs your actual script..command.sh: The script block you wrote in your Nextflow process—contains the actual scientific commands (e.g., running FastQC).
Summary:
-
.command.shis your pipeline logic. -
.command.runis Nextflow’s automation wrapper. -
.command.outand.command.errhelp with debugging. -
All files together provide a complete audit trail for reproducibility and troubleshooting. Complete example folder structure
drwxrwxr-x 2 user user 4096 Feb 18 15:32 .
drwxrwxr-x 3 user user 4096 Feb 18 15:32 ..
-rw-rw-r-- 1 user user 0 Feb 18 15:32 .command.begin
-rw-rw-r-- 1 user user 1750 Feb 18 15:32 .command.err
-rw-rw-r-- 1 user user 1868 Feb 18 15:32 .command.log
-rw-rw-r-- 1 user user 118 Feb 18 15:32 .command.out
-rw-rw-r-- 1 user user 3758 Feb 18 15:32 .command.run ← Wrapper script
-rw-rw-r-- 1 user user 347 Feb 18 15:32 .command.sh ← Your actual script
-rw-rw-r-- 1 user user 1 Feb 18 15:32 .exitcode
-rw-rw-r-- 1 user user 598807 Feb 18 15:32 sample1_R1_fastqc.html
-rw-rw-r-- 1 user user 352734 Feb 18 15:32 sample1_R1_fastqc.zip
lrwxrwxrwx 1 user user 113 Feb 18 15:32 sample1_R1.fastq.gz -> /path/to/data/sample1_R1.fastq.gz
-rw-rw-r-- 1 user user 582761 Feb 18 15:32 sample1_R2_fastqc.html
-rw-rw-r-- 1 user user 326969 Feb 18 15:32 sample1_R2_fastqc.zip
lrwxrwxrwx 1 user user 113 Feb 18 15:32 sample1_R2.fastq.gz -> /path/to/data/sample1_R2.fastq.gz
-rw-rw-r-- 1 user user 50 Feb 18 15:32 versions.yml
sample1_R2.fastq.gz -> /path/to/data/sample1_R2.fastq.gz:
- By default, for each step, to reduce the disk usages, we can simply create a
symlinkfrom previous step. - It can redirect the output from previous step to use in the current folder instead of copying again that causes the duplication
- For cloud environment, it has a challege is that it will download the file inputs in each process in each machine instead of sharing file system with high I/O speed
You can view the .command.run
#!/bin/bash
### ---
### name: 'FASTQC (1)'
### outputs:
### - '*.html'
### - '*.zip'
### ...
set -e
set -u
NXF_DEBUG=${NXF_DEBUG:=0}; [[ $NXF_DEBUG > 1 ]] && set -x
NXF_ENTRY=${1:-nxf_main}
# Sleep for a given number of seconds (fallback to 1s if error)
nxf_sleep() {
sleep $1 2>/dev/null || sleep 1;
}
# Generate a timestamp in milliseconds
nxf_date() {
local ts=$(date +%s%3N);
if [[ ${#ts} == 10 ]]; then echo ${ts}000
elif [[ $ts == *%3N ]]; then echo ${ts/\%3N/000}
elif [[ $ts == *3N ]]; then echo ${ts/3N/000}
elif [[ ${#ts} == 13 ]]; then echo $ts
else echo "Unexpected timestamp value: $ts"; exit 1
fi
}
# Print sorted environment variables, masking AWS credentials
nxf_env() {
echo '============= task environment ============='
env | sort | sed "s/\(.*\)AWS\(.*\)=\(.\{6\}\).*/\1AWS\2=\3xxxxxxxxxxxxx/"
echo '============= task output =================='
}
# Kill a process and all its child processes
nxf_kill() {
declare -a children
while read P PP;do
children[$PP]+=" $P"
done < <(ps -e -o pid= -o ppid=)
kill_all() {
[[ $1 != $$ ]] && kill $1 2>/dev/null || true
for i in ${children[$1]:=}; do kill_all $i; done
}
kill_all $1
}
# Create a temporary directory
nxf_mktemp() {
local base=${1:-/tmp}
mkdir -p "$base"
if [[ $(uname) = Darwin ]]; then mktemp -d $base/nxf.XXXXXXXXXX
else TMPDIR="$base" mktemp -d -t nxf.XXXXXXXXXX
fi
}
# Copy files/directories recursively, preserving symlinks
nxf_fs_copy() {
local source=$1
local target=$2
local basedir=$(dirname $1)
mkdir -p $target/$basedir
cp -fRL $source $target/$basedir
}
# Move files/directories recursively
nxf_fs_move() {
local source=$1
local target=$2
local basedir=$(dirname $1)
mkdir -p $target/$basedir
mv -f $source $target/$basedir
}
# Rsync files/directories recursively
nxf_fs_rsync() {
rsync -rRl $1 $2
}
# Copy files using rclone
nxf_fs_rclone() {
rclone copyto $1 $2/$1
}
# Copy files using fcp
nxf_fs_fcp() {
fcp $1 $2/$1
}
# Handler for process exit: sets exit code and exits
on_exit() {
local last_err=$?
local exit_status=${nxf_main_ret:=0}
[[ ${exit_status} -eq 0 && ${nxf_unstage_ret:=0} -ne 0 ]] && exit_status=${nxf_unstage_ret:=0}
[[ ${exit_status} -eq 0 && ${last_err} -ne 0 ]] && exit_status=${last_err}
printf -- $exit_status > /home/giangnguyen/Documents/dev/nf-germline-short-read-variant-calling/tmp/work/4b/957bdd32e8e03d575e17185ef4f09a/.exitcode
set +u
exit $exit_status
}
# Handler for termination signals: kills child processes
on_term() {
set +e
[[ "$pid" ]] && nxf_kill $pid
}
# Launch the main command script
nxf_launch() {
/bin/bash -ue /home/giangnguyen/Documents/dev/nf-germline-short-read-variant-calling/tmp/work/4b/957bdd32e8e03d575e17185ef4f09a/.command.sh
}
# Stage input files (create symlinks in work directory)
nxf_stage() {
true
# stage input files
rm -f test_1.fastq.gz
rm -f test_2.fastq.gz
ln -s /home/giangnguyen/Documents/dev/nf-germline-short-read-variant-calling/tmp/work/stage-8a00ef45-80ce-4033-9494-354f1e68968c/a7/c6160733f5332acde30841c8d7cfb6/test_1.fastq.gz test_1.fastq.gz
ln -s /home/giangnguyen/Documents/dev/nf-germline-short-read-variant-calling/tmp/work/stage-8a00ef45-80ce-4033-9494-354f1e68968c/2e/f8faa73d537d3b8bc792454dc541de/test_2.fastq.gz test_2.fastq.gz
}
# Unstage output files (placeholder, does nothing)
nxf_unstage_outputs() {
true
}
# Unstage control files (placeholder, does nothing)
nxf_unstage_controls() {
true
}
# Unstage outputs and controls after main execution
nxf_unstage() {
if [[ ${nxf_main_ret:=0} == 0 ]]; then
(set -e -o pipefail; (nxf_unstage_outputs | tee -a .command.out) 3>&1 1>&2 2>&3 | tee -a .command.err)
nxf_unstage_ret=$?
fi
nxf_unstage_controls
}
# Main entry point: sets up traps, environment, stages files, runs command, unstages outputs
nxf_main() {
trap on_exit EXIT
trap on_term TERM INT USR2
trap '' USR1
[[ "${NXF_CHDIR:-}" ]] && cd "$NXF_CHDIR"
NXF_SCRATCH=''
[[ $NXF_DEBUG > 0 ]] && nxf_env
touch /home/giangnguyen/Documents/dev/nf-germline-short-read-variant-calling/tmp/work/4b/957bdd32e8e03d575e17185ef4f09a/.command.begin
set +u
set -u
[[ $NXF_SCRATCH ]] && cd $NXF_SCRATCH
export NXF_TASK_WORKDIR="$PWD"
nxf_stage
set +e
(set -o pipefail; (nxf_launch | tee .command.out) 3>&1 1>&2 2>&3 | tee .command.err) &
pid=$!
wait $pid || nxf_main_ret=$?
nxf_unstage
}
# Run the entry point (default: nxf_main)
$NXF_ENTRY
Overall, the above script will:
- Download data
- Run the command:
/bin/bash -ue /home/giangnguyen/Documents/dev/nf-germline-short-read-variant-calling/tmp/work/4b/957bdd32e8e03d575e17185ef4f09a/.command.sh - Output the data
3.4 Run Workflow With Sigularity Container
- Singularity or any container is the technology that helps to have the reproducible environment.
- It is portable which means you can generate the environment, upload to elsewhere, download to use in any machine if it has the container setting
- Better with this blog: containers-hpc-docker-singularity-apptainer
As mentioned above, we can add singularity configuration in the template
#!/usr/bin/env nextflow
nextflow.enable.dsl=2 // enable DSL2
// Define the module as template
process FASTQC {
container 'quay.io/biocontainers/fastqc:0.12.1--hdfd78af_0' // NEW CONFIG
input:
tuple val(meta), path(reads) // input variable, example: [sample, [/path/read1.fq, /path/read2.fq ]]
output:
tuple val(meta), path("*.html") // output variable, example: [sample, [/path/read1.html, /path/read2.html ]]
script:
"""
# Quality Control with FastQC
# This generates HTML and ZIP files with quality metrics
fastqc $reads
"""
}
workflow {
ch_input = Channel.of([ params.sample , [file(params.fastq_r1), file(params.fastq_r2)] ])
// Run workflow
FASTQC (
ch_input
)
}
3.5. Run Nextflow Pipeline (With Singularity)
Create the file nextflow.config on the same folder
singularity.enabled = true
singularity.autoMounts = true
nextflow run main.nf -resume \
--sample sample1 \
--fastq_r1 https://raw.githubusercontent.com/nf-core/test-datasets/modules/data/genomics/homo_sapiens/illumina/fastq/test_1.fastq.gz \
--fastq_r2 https://raw.githubusercontent.com/nf-core/test-datasets/modules/data/genomics/homo_sapiens/illumina/fastq/test_2.fastq.gz
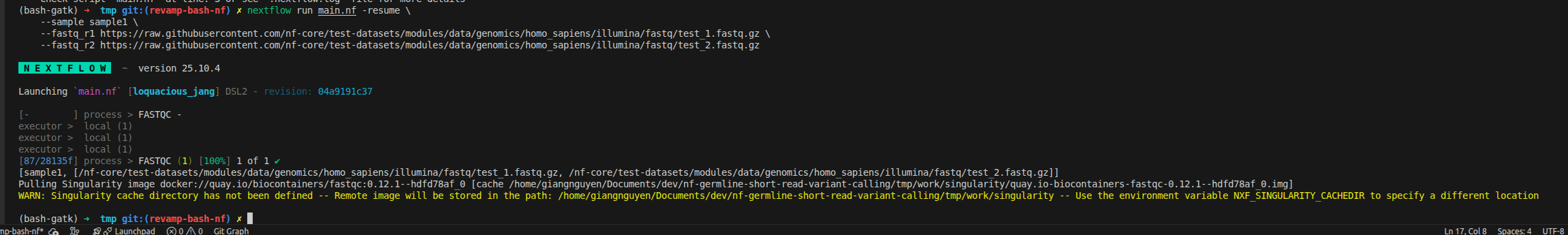
What happens:
- Nextflow reads the
containerdirective fromfastqc.nf - Pulls Docker image from
quay.io/biocontainers/fastqc:0.12.1--hdfd78af_0 - Converts to Singularity
.imgformat (cached inwork/singularity/) - Runs FastQC inside the container (no root privileges required)
We can validate how it work by seeing the update on the .command.run
nxf_launch() {
set +u; env - PATH="$PATH" ${TMP:+SINGULARITYENV_TMP="$TMP"} ${TMPDIR:+SINGULARITYENV_TMPDIR="$TMPDIR"} ${NXF_TASK_WORKDIR:+SINGULARITYENV_NXF_TASK_WORKDIR="$NXF_TASK_WORKDIR"} singularity exec --no-home --pid -B /home/giangnguyen/Documents/dev/nf-germline-short-read-variant-calling/tmp/work /home/giangnguyen/Documents/dev/nf-germline-short-read-variant-calling/tmp/work/singularity/quay.io-biocontainers-fastqc-0.12.1--hdfd78af_0.img /bin/bash -c "cd $NXF_TASK_WORKDIR; /bin/bash -ue /home/giangnguyen/Documents/dev/nf-germline-short-read-variant-calling/tmp/work/87/28135f97ffa37daf6192aaf824dedb/.command.sh"
}
3.6 Run With Memory And Cpus Limit (With Docker)
- Running without setup the cluster/batch executor, singularity/apptainer does not support limit CPUs and memory
- Using
dockerinstead - Not only docker, nextflow can be expanded to support more container engine using simple nextflow config
- Almost, it will be based on the docker with many well known register (quay.io, docker.io) with extensive images that can converted to their own specific container
#!/usr/bin/env nextflow
nextflow.enable.dsl=2 // enable DSL2
// Define the module as template
process FASTQC {
container 'quay.io/biocontainers/fastqc:0.12.1--hdfd78af_0'
cpus 2 // NEW CONFIG
memory "4.GB" // NEW CONFIG
input:
tuple val(meta), path(reads) // input variable, example: [sample, [/path/read1.fq, /path/read2.fq ]]
output:
tuple val(meta), path("*.html") // output variable, example: [sample, [/path/read1.html, /path/read2.html ]]
script:
"""
# Quality Control with FastQC
# This generates HTML and ZIP files with quality metrics
fastqc $reads
"""
}
workflow {
ch_input = Channel.of([ params.sample , [file(params.fastq_r1), file(params.fastq_r2)] ])
// Run workflow
FASTQC (
ch_input
)
}
You can keep the nextlfow.config, there is no change at all on the singularity command
Change nextflow.config to use docker
docker.enabled = true
docker.autoMounts = true
Checking the command to see how docker add config to limit cpus and memory
# check docker command quickly
cat work/8e/7c7d8e091a9b515adbe0f136e13f93/.command.run|grep docker
The below statement shows clearly it uses docker with limit resources --cpu-shares 2048 --memory 4096m
docker run -i --cpu-shares 2048 --memory 4096m -e "NXF_TASK_WORKDIR" -v /home/giangnguyen/Documents/dev/nf-germline-short-read-variant-calling/tmp/work:/home/giangnguyen/Documents/dev/nf-germline-short-read-variant-calling/tmp/work -w "$NXF_TASK_WORKDIR" --name $NXF_BOXID quay.io/biocontainers/fastqc:0.12.1--hdfd78af_0 /bin/bash -ue /home/giangnguyen/Documents/dev/nf-germline-short-read-variant-calling/tmp/work/8e/7c7d8e091a9b515adbe0f136e13f93/.command.sh
Remember to clean your work directory, changing container, cpus and memory does not change the process hash, which caches existing results
3.7 Run With Samplesheet
For pipeline, the raw data is usually defined by a csv file called samplesheet. Inside, we have a logic to read the csv file and collect the channels
#!/usr/bin/env nextflow
nextflow.enable.dsl=2 // enable DSL2
// Define the module as template
process FASTQC {
container 'quay.io/biocontainers/fastqc:0.12.1--hdfd78af_0'
cpus 2 // NEW CONFIG
memory "4.GB" // NEW CONFIG
input:
tuple val(meta), path(reads) // input variable, example: [sample, [/path/read1.fq, /path/read2.fq ]]
output:
tuple val(meta), path("*.html") // output variable, example: [sample, [/path/read1.html, /path/read2.html ]]
script:
"""
# Quality Control with FastQC
# This generates HTML and ZIP files with quality metrics
fastqc $reads
"""
}
workflow {
Channel
.fromPath(params.input)
.splitCsv(header: true)
.map { row ->
def meta = [:] // create empty list for metadata
meta.id = row.sample // it map value of row as sample id, add to meta list
// Support both new format (with lane) and old format (without lane)
meta.lane = row.lane // require to have lane, for combining data later if one sample is sequenced by more than one lanes
def reads = [] // create empty list for files
reads.add(file(row.fastq_1)) // add R1 fq
reads.add(file(row.fastq_2)) // add R2 fq
return [ meta, reads ]
}
.set { ch_input }
ch_input.view() // View channel here
// Run workflow
FASTQC (
ch_input
)
}
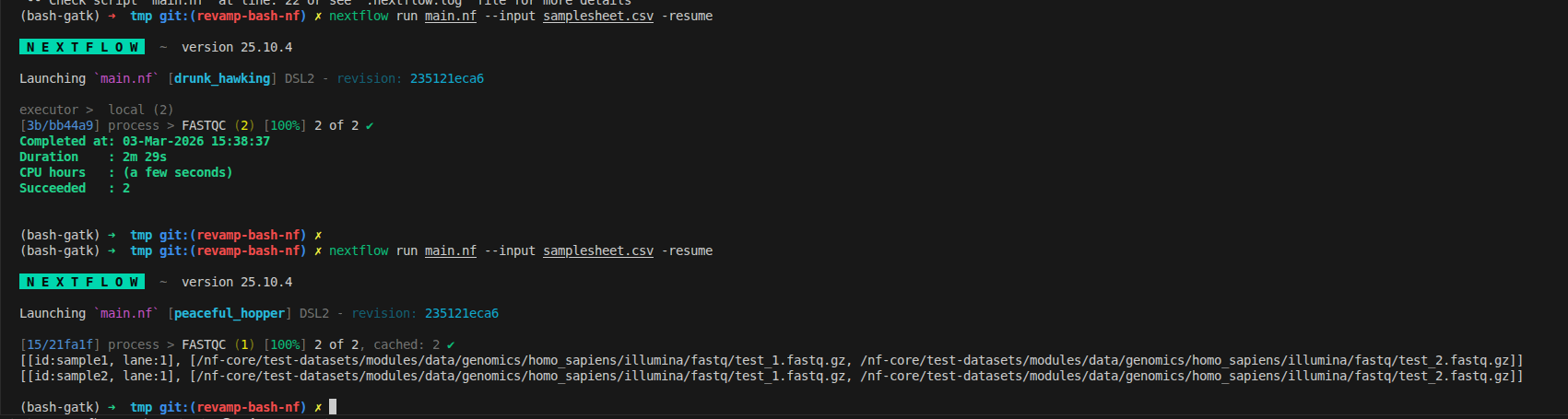
We can rerun with the input is samplesheet.csv content as below
sample,lane,fastq_1,fastq_2
sample1,1,https://raw.githubusercontent.com/nf-core/test-datasets/modules/data/genomics/homo_sapiens/illumina/fastq/test_1.fastq.gz,https://raw.githubusercontent.com/nf-core/test-datasets/modules/data/genomics/homo_sapiens/illumina/fastq/test_2.fastq.gz
sample2,1,https://raw.githubusercontent.com/nf-core/test-datasets/modules/data/genomics/homo_sapiens/illumina/fastq/test_1.fastq.gz,https://raw.githubusercontent.com/nf-core/test-datasets/modules/data/genomics/homo_sapiens/illumina/fastq/test_2.fastq.gz
We can rerun and see how that there are 2 tuples which matches with the input file. From now, it is the true meaning of distribution tasks, running 2 FASTQC at the same time on my machine, has the resource limitation via docker
nextflow run main.nf --input samplesheet.csv -resume
3.8 Connect modules
A pipeline is a chain of tools working together, where each step’s output feeds into the next. Let’s extend our example by adding trimgalore and bwa modules.
What’s new?
- Two new modules:
trimgalore(for adapter trimming) andbwa(for alignment). trimgaloreruns directly onch_inputand emits its output asreads.- The output from
trimgalore(TRIM_GALORE.out.reads) is used as the input forbwa, maintaining the correct tuple format. - You can specify which output to use from a process by referencing
.out.<emit_name>. - The
scriptblock can includedefstatements to define variables dynamically. - When creating channels for reference files, use
.collect()to allow reuse across multiple processes (since channels are queues by default).
The below structure demonstrates how to chain processes, pass outputs between modules, and manage shared resources in Nextflow. :::
#!/usr/bin/env nextflow
nextflow.enable.dsl=2 // enable DSL2
// Define the module as template
process FASTQC {
container 'quay.io/biocontainers/fastqc:0.12.1--hdfd78af_0'
cpus 2 // NEW CONFIG
memory "4.GB" // NEW CONFIG
input:
tuple val(meta), path(reads) // input variable, example: [sample, [/path/read1.fq, /path/read2.fq ]]
output:
tuple val(meta), path("*.html") // output variable, example: [sample, [/path/read1.html, /path/read2.html ]]
script:
"""
# Quality Control with FastQC
# This generates HTML and ZIP files with quality metrics
fastqc $reads
"""
}
process TRIM_GALORE {
container 'quay.io/biocontainers/trim-galore:0.6.10--hdfd78af_0'
cpus 2 // NEW CONFIG
memory "4.GB" // NEW CONFIG
input:
tuple val(meta), path(reads)
output:
tuple val(meta), path("*_val_{1,2}.fq.gz"), emit: reads
tuple val(meta), path("*_trimming_report.txt"), emit: log
script:
"""
# Adapter Trimming and Quality Filtering
trim_galore \\
--paired \\
--quality 20 --length 50 --fastqc \\
--cores $task.cpus \\
${reads}
"""
}
process BWA_MEM {
container 'quay.io/biocontainers/mulled-v2-fe8faa35dbf6dc65a0f7f5d4ea12e31a79f73e40:219b6c272b25e7e642ae3ff0bf0c5c81a5135ab4-0'
cpus 2 // NEW CONFIG
memory "4.GB" // NEW CONFIG
input:
tuple val(meta), path(reads)
path reference
path reference_fai
path reference_dict
path bwa_index
output:
tuple val(meta), path("*.bam"), emit: bam
script:
def args = task.ext.args ?: '-M'
def prefix = task.ext.prefix ?: "${meta.id}"
def read_group = "@RG\\tID:${meta.id}\\tSM:${meta.id}\\tPL:ILLUMINA\\tLB:${meta.id}_lib"
"""
# Read Alignment with BWA-MEM
bwa mem \\
-t $task.cpus \\
$args \\
-R "${read_group}" \\
$reference \\
${reads[0]} ${reads[1]} \\
| samtools view -Sb - > ${prefix}_aligned.bam
"""
}
workflow {
Channel
.fromPath(params.input)
.splitCsv(header: true)
.map { row ->
def meta = [:] // create empty list for metadata
meta.id = row.sample // it map value of row as sample id, add to meta list
// Support both new format (with lane) and old format (without lane)
meta.lane = row.lane // require to have lane, for combining data later if one sample is sequenced by more than one lanes
def reads = [] // create empty list for files
reads.add(file(row.fastq_1)) // add R1 fq
reads.add(file(row.fastq_2)) // add R2 fq
return [ meta, reads ]
}
.set { ch_input }
// Run workflow
FASTQC (
ch_input
)
TRIM_GALORE (
ch_input
)
ch_reference = Channel.fromPath(params.reference).collect()
ch_reference_fai = Channel.fromPath(params.reference + ".fai").collect()
ch_reference_dict = Channel.fromPath(params.reference_dict).collect()
ch_bwa_index = Channel.fromPath(params.bwa_index+".{amb,ann,bwt,pac,sa}").collect()
BWA_MEM (
TRIM_GALORE.out.reads, // use output of trim_galore as input for bwa_mem
ch_reference,
ch_reference_fai,
ch_reference_dict,
ch_bwa_index
)
}
We can rerun to ensure it work perfectly now
nextflow run main.nf --input samplesheet.csv -resume
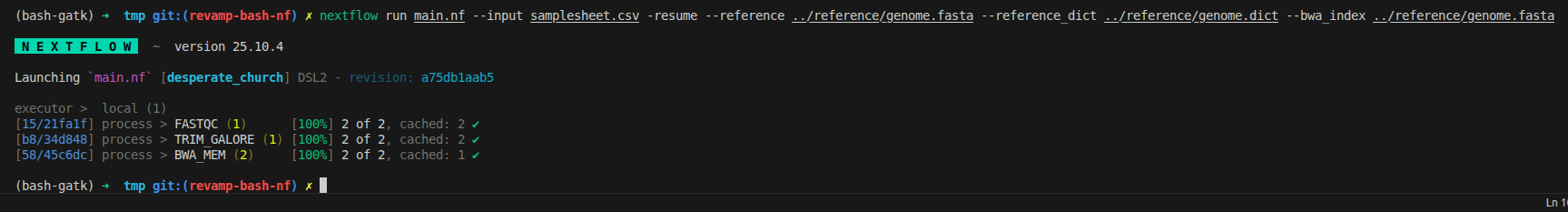
3.9 Reorganize workflow
The final nextflow version only include:
- Organized modules
- Single
nextflow.config - Single
main.nfonly import and usemodules - More additional advances will be configured later on next blog
3.9.1 Modules/Subworkflows
This one shows you how to split your single script into modules/subworkflows that can be easier for management.
For modules, you can create a folder called modules then import it as a library/package that can be reused later on your main.nf
// Include modules
include { FASTQC } from './modules/fastqc'
include { TRIM_GALORE } from './modules/trim_galore'
include { BWA_MEM } from './modules/bwa_mem'
include { SAMTOOLS_SORT } from './modules/samtools_sort'
include { GATK_MARKDUPLICATES } from './modules/gatk_markduplicates'
include { GATK_BASERECALIBRATOR } from './modules/gatk_baserecalibrator'
include { GATK_APPLYBQSR } from './modules/gatk_applybqsr'
include { GATK_COLLECTMETRICS } from './modules/gatk_collectmetrics'
include { GATK_HAPLOTYPECALLER } from './modules/gatk_haplotypecaller'
include { GATK_GENOTYPEGVCFS } from './modules/gatk_genotypegvcfs'
For subworkflow, it is stack of many modules. Inside, we have a combination of tools, have a specific name for workflow to import later, has input and output similar to the process
#!/usr/bin/env nextflow
/*
========================================================================================
PREPROCESSING SUBWORKFLOW
========================================================================================
Steps 1-8: QC, Trimming, Alignment, Deduplication, BQSR, Quality Metrics
========================================================================================
*/
include { FASTP } from '../../../modules/local/fastp/trim/main'
include { BWAMEM2_INDEX } from '../../../modules/local/bwa/index/main'
include { BWA_MEM2 } from '../../../modules/local/bwa/mem2/main'
include { SAMTOOLS_SORT } from '../../../modules/local/samtools/sort/main'
include { SAMTOOLS_MERGE } from '../../../modules/local/samtools/merge/main'
include { GATKSPARK_MARKDUPLICATES } from '../../../modules/local/gatkspark/markduplicates/main'
include { GATKSPARK_BASERECALIBRATOR } from '../../../modules/local/gatkspark/baserecalibrator/main'
include { GATKSPARK_APPLYBQSR } from '../../../modules/local/gatkspark/applybqsr/main'
include { GATK_COLLECTMETRICS } from '../../../modules/local/gatk/collectmetrics/main'
workflow PREPROCESSING {
take:
reads_ch // channel: [ val(meta), [ path(read1), path(read2) ] ]
ref_fasta // path: reference FASTA
ref_fai // path: reference FAI
ref_dict // path: reference dict
bwa2_index // channel: Optional BWA index files
index_bwa2_reference // channel: Optional BWA index files
dbsnp_vcf // path: dbSNP VCF
dbsnp_tbi // path: dbSNP TBI
known_indels_vcf // path: known indels VCF
known_indels_tbi // path: known indels TBI
main:
ch_versions = Channel.empty()
// Check if BWA index needs to be generated
// If empty, generate it; otherwise use provided index
if (index_bwa2_reference){
BWAMEM2_INDEX(ref_fasta)
ch_versions = ch_versions.mix(BWAMEM2_INDEX.out.versions)
bwa2_index_ch = BWAMEM2_INDEX.out.index
}
else{
bwa2_index_ch = Channel.fromPath(bwa2_index)
}
//
// STEP 1: Adapter Trimming, Quality Filtering, and QC with fastp
//
FASTP (
reads_ch
)
ch_versions = ch_versions.mix(FASTP.out.versions)
//
// STEP 2: Read Alignment with BWA-MEM2
//
BWA_MEM2 (
FASTP.out.reads,
ref_fasta,
ref_fai,
ref_dict,
bwa2_index_ch
)
ch_versions = ch_versions.mix(BWA_MEM2.out.versions)
//
// STEP 3: Sort BAM file
//
SAMTOOLS_SORT (
BWA_MEM2.out.bam
)
ch_versions = ch_versions.mix(SAMTOOLS_SORT.out.versions)
//
// STEP 4: Merge lanes per sample (if multiple lanes exist)
//
SAMTOOLS_SORT.out.bam
.map { meta, bam ->
def new_meta = [:]
new_meta.id = meta.sample
new_meta.sample = meta.sample
return [ new_meta.id, new_meta, bam ]
}
.groupTuple()
.map { sample_id, metas, bams ->
return [ metas[0], bams ]
}
.set { ch_bams_to_merge }
SAMTOOLS_MERGE (
ch_bams_to_merge
)
ch_versions = ch_versions.mix(SAMTOOLS_MERGE.out.versions)
//
// STEP 5: Mark Duplicates with GATK Spark
//
GATKSPARK_MARKDUPLICATES (
SAMTOOLS_MERGE.out.bam
)
ch_versions = ch_versions.mix(GATKSPARK_MARKDUPLICATES.out.versions)
//
// STEP 6: Base Quality Score Recalibration - Generate table
//
GATKSPARK_BASERECALIBRATOR (
GATKSPARK_MARKDUPLICATES.out.bam.join(GATKSPARK_MARKDUPLICATES.out.bai),
ref_fasta,
ref_fai,
ref_dict,
dbsnp_vcf,
dbsnp_tbi,
known_indels_vcf,
known_indels_tbi
)
ch_versions = ch_versions.mix(GATKSPARK_BASERECALIBRATOR.out.versions)
//
// STEP 7: Apply BQSR
//
GATKSPARK_APPLYBQSR (
GATKSPARK_MARKDUPLICATES.out.bam
.join(GATKSPARK_MARKDUPLICATES.out.bai)
.join(GATKSPARK_BASERECALIBRATOR.out.table),
ref_fasta,
ref_fai,
ref_dict
)
ch_versions = ch_versions.mix(GATKSPARK_APPLYBQSR.out.versions)
//
// STEP 8: Collect Alignment Metrics
//
GATK_COLLECTMETRICS (
GATKSPARK_APPLYBQSR.out.bam.join(GATKSPARK_APPLYBQSR.out.bai),
ref_fasta,
ref_fai,
ref_dict
)
ch_versions = ch_versions.mix(GATK_COLLECTMETRICS.out.versions)
emit:
bam = GATKSPARK_APPLYBQSR.out.bam // channel: [ val(meta), path(bam) ]
bai = GATKSPARK_APPLYBQSR.out.bai // channel: [ val(meta), path(bai) ]
alignment_summary = GATK_COLLECTMETRICS.out.alignment_summary // channel: [ val(meta), path(metrics) ]
insert_metrics = GATK_COLLECTMETRICS.out.insert_metrics // channel: [ val(meta), path(metrics) ]
versions = ch_versions // channel: path(versions.yml)
}
In the main.nf, it can be imported as module
include { PREPROCESSING } from './subworkflows/local/preprocessing/main'
3.9.2 Config And Profiles
We currently have only 2 lines on the config
docker.enabled = true
docker.autoMounts = true
We migrate to below script where:
- We can define the params directly inside in
params {} - We can split the config to smaller config
profileis the concept that we collect a set of parameters/setup to run that can be easier for configuration by adding-profile test,dockerto run withtestparameters collection anddockerconfigurationmanifestis good to have to show manifest of the pipelinetimeline,traceandreportshould be enable that helps to run with better monitoringcheck_maxandlabelhelp to configure cpus and memory properly. If we have only 4 CPUs, running only 2 FASTQC processed are allows while running more processes will cause the issue of resource allocation
/*
========================================================================================
GATK Variant Calling Pipeline - Nextflow Config
========================================================================================
*/
// Global default params
params {
// Input/output options
input = null
outdir = 'results_nextflow'
// Direct input parameters (alternative to samplesheet)
sample = 'sample1'
fastq_r1 = null
fastq_r2 = null
// Reference files
reference = '../reference/genome.fasta'
dbsnp = '../reference/dbsnp_146.hg38.vcf.gz'
known_indels = '../reference/mills_and_1000G.indels.vcf.gz'
// Resource limits
max_cpus = 4
max_memory = '15.GB'
max_time = '24.h'
// Help
help = false
}
// Modules
process {
publishDir = [
path: { "${params.outdir}/${task.process.toLowerCase()}" },
mode: "symlink",
enabled: true
]
}
// Process configuration
process {
// Default settings
shell = ['/bin/bash', '-euo', 'pipefail']
// Error handling
errorStrategy = { task.exitStatus in [143,137,104,134,139] ? 'retry' : 'finish' }
maxRetries = 1
maxErrors = '-1'
// Process labels
withLabel: process_low {
cpus = { check_max( 2 * task.attempt, 'cpus' ) }
memory = { check_max( 2.GB * task.attempt, 'memory' ) }
time = { check_max( 4.h * task.attempt, 'time' ) }
}
withLabel: process_medium {
cpus = { check_max( 4 * task.attempt, 'cpus' ) }
memory = { check_max( 4.GB * task.attempt, 'memory' ) }
time = { check_max( 8.h * task.attempt, 'time' ) }
}
withLabel: process_high {
cpus = { check_max( 4 * task.attempt, 'cpus' ) }
memory = { check_max( 16.GB * task.attempt, 'memory' ) }
time = { check_max( 16.h * task.attempt, 'time' ) }
}
}
// Execution profiles
profiles {
docker {
docker.enabled = true
docker.userEmulation = true
singularity.enabled = false
podman.enabled = false
shifter.enabled = false
charliecloud.enabled = false
}
singularity {
singularity.enabled = true
singularity.autoMounts = true
docker.enabled = false
podman.enabled = false
shifter.enabled = false
charliecloud.enabled = false
}
test {
params.sample = 'sample1'
params.fastq_r1 = '../data/sample1_R1.fastq.gz'
params.fastq_r2 = '../data/sample1_R2.fastq.gz'
params.reference = '../reference/genome.fasta'
params.dbsnp = '../reference/dbsnp_146.hg38.vcf.gz'
params.known_indels = '../reference/mills_and_1000G.indels.vcf.gz'
params.max_cpus = 4
params.max_memory = '15.GB'
}
}
// Manifest
manifest {
name = 'GATK Variant Calling Pipeline'
author = 'River'
homePage = 'https://github.com/your-repo/gatk-variant-calling'
description = 'GATK best practices variant calling pipeline'
mainScript = 'main.nf'
nextflowVersion = '!>=22.10.0'
version = '1.0.0'
}
// Function to ensure that resource requirements don't go beyond maximum limit
def check_max(obj, type) {
if (type == 'memory') {
try {
if (obj.compareTo(params.max_memory as nextflow.util.MemoryUnit) == 1)
return params.max_memory as nextflow.util.MemoryUnit
else
return obj
} catch (all) {
println "WARNING: Max memory '${params.max_memory}' is not valid! Using default value: $obj"
return obj
}
} else if (type == 'time') {
try {
if (obj.compareTo(params.max_time as nextflow.util.Duration) == 1)
return params.max_time as nextflow.util.Duration
else
return obj
} catch (all) {
println "WARNING: Max time '${params.max_time}' is not valid! Using default value: $obj"
return obj
}
} else if (type == 'cpus') {
try {
return Math.min( obj, params.max_cpus as int )
} catch (all) {
println "WARNING: Max cpus '${params.max_cpus}' is not valid! Using default value: $obj"
return obj
}
}
}
On the modules, now it can be updated with:
- tag: Instead of number, it show you on the terminal which step is running via tag
- label: Automatically define the cpus and memory. I don't like this too much in the industry application. We should optimize to have around 80% of resource usage
- version: Version tracking is nessesary, it can be apply with
MULTIQCto generate the report that has all tools version that we do not manually tracking on each process
process FASTQC {
tag "$meta.id"
label 'process_medium'
// Use BioContainers FastQC image
// Works with both Docker and Singularity (no root required)
container 'quay.io/biocontainers/fastqc:0.12.1--hdfd78af_0'
input:
tuple val(meta), path(reads)
output:
tuple val(meta), path("*.html"), emit: html
tuple val(meta), path("*.zip") , emit: zip
path "versions.yml" , emit: versions
when:
task.ext.when == null || task.ext.when
script:
def args = task.ext.args ?: ''
def prefix = task.ext.prefix ?: "${meta.id}"
// Determine if single-end or paired-end
def input_files = reads instanceof List ? reads.join(' ') : reads
"""
# Quality Control with FastQC
# This generates HTML and ZIP files with quality metrics
fastqc \\
$args \\
--threads $task.cpus \\
$input_files
# Create versions file
cat <<-END_VERSIONS > versions.yml
"${task.process}":
fastqc: \$(fastqc --version | sed 's/FastQC v//')
END_VERSIONS
"""
}
To publish directory, using the below config is much easier with output folder is lowercase for all processes
process {
publishDir = [
path: { "${params.outdir}/${task.process.toLowerCase()}" },
mode: "symlink",
enabled: true
]
}
3.10 Final Nextflow Workflow
Now you can have the final workflow that can be ensure you understand the standard concept of nextflow. You need to run the download script first that help to prepare the input and reference data.
git clone git@github.com:nttg8100/nf-germline-short-read-variant-calling.git -b 0.1.0
cd nf-germline-short-read-variant-calling
# Install dependencies
pixi shell --manifest-path bash-gatk
# Download test data
bash scripts/download_data.sh
# Run pipeline
cd nextflow-gatk
nextflow run main.nf -profile docker
Rerun to get the final workflow
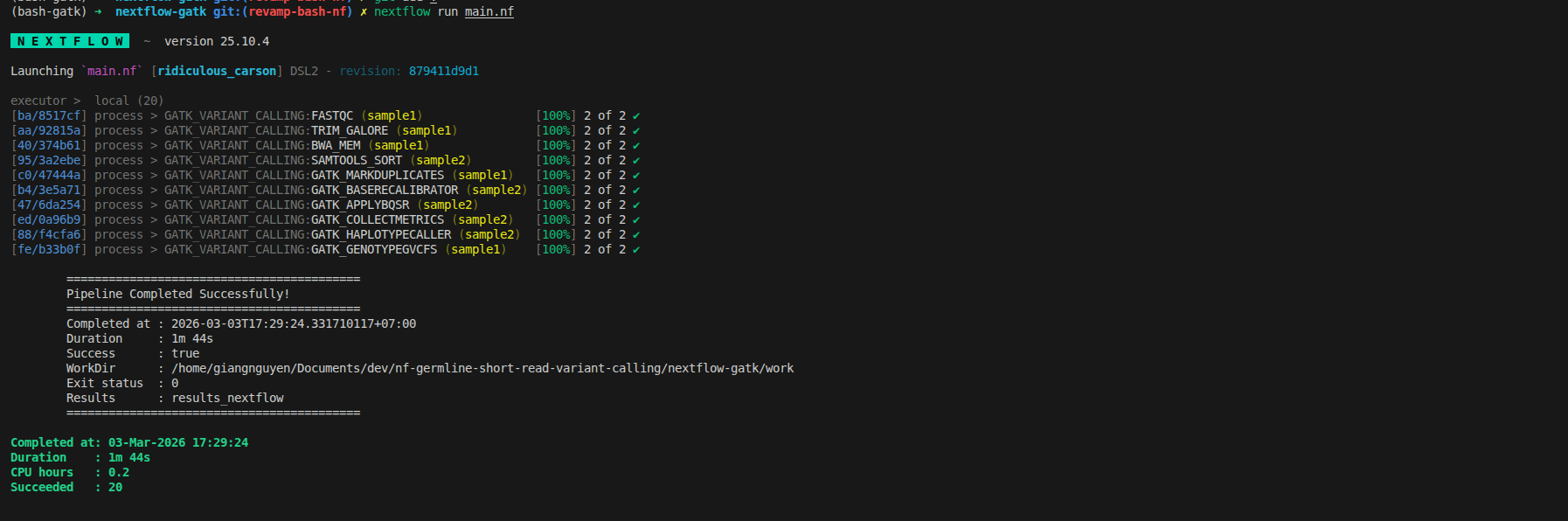
4. Validate migration
The simple idea is that it may run with different controller (bash vs nextflow), the result should be consistent. To convert it successfully, here is the tip
- Using md5sum to ensure the content are the same
- Ensure the tool generate result consistently by itself, running bash workflow twice it should have the same output files
- Removing the different factors: timestamp in zip, random calculation on tools
With FASTQC, first step, we can remove the timestamp in zip file, then compare the content of file inside
#!/bin/bash
#
# Validation Script: Compare Bash and Nextflow Outputs
# This script validates that the Nextflow pipeline produces identical results to the bash pipeline
#
set -euo pipefail
echo "=========================================="
echo "Pipeline Migration Validation"
echo "Comparing Bash vs Nextflow (Step 1: FastQC)"
echo "=========================================="
echo
# Directories
BASH_DIR="bash-gatk/results/qc/sample1"
NEXTFLOW_DIR="nextflow-gatk/results_nextflow/qc/sample1"
echo "=== File Comparison ==="
echo
# Compare all HTML files
for file in ${BASH_DIR}/*.html; do
fname=$(basename "$file")
if [[ -f "${NEXTFLOW_DIR}/$fname" ]]; then
BASH_MD5=$(md5sum "$file" | cut -d' ' -f1)
NEXTFLOW_MD5=$(md5sum "${NEXTFLOW_DIR}/$fname" | cut -d' ' -f1)
if [ "$BASH_MD5" == "$NEXTFLOW_MD5" ]; then
echo " ✓ PASS: $fname"
echo " MD5: $BASH_MD5"
else
echo " ✗ FAIL: $fname"
echo " Bash MD5: $BASH_MD5"
echo " Nextflow MD5: $NEXTFLOW_MD5"
fi
else
echo " ✗ FAIL: $fname (missing in Nextflow)"
fi
done
echo
echo "Comparing ZIP files (content only, ignoring timestamps):"
# Compare all ZIP files (ignore archive timestamps, check actual content)
for file in ${BASH_DIR}/*.zip; do
fname=$(basename "$file")
if [[ -f "${NEXTFLOW_DIR}/$fname" ]]; then
BASE_NAME=$(basename "$file" .zip)
BASH_EXTRACT="/tmp/bash_${BASE_NAME}"
NF_EXTRACT="/tmp/nextflow_${BASE_NAME}"
rm -rf "$BASH_EXTRACT" "$NF_EXTRACT"
mkdir -p "$BASH_EXTRACT" "$NF_EXTRACT"
unzip -q "$file" -d "$BASH_EXTRACT"
unzip -q "${NEXTFLOW_DIR}/$fname" -d "$NF_EXTRACT"
if diff -r "$BASH_EXTRACT" "$NF_EXTRACT" > /dev/null 2>&1; then
echo " ✓ PASS: $fname (content identical)"
else
echo " ✗ FAIL: $fname (content differs)"
fi
FASTQC_DATA="${BASE_NAME}/fastqc_data.txt"
if [ -f "$BASH_EXTRACT/$FASTQC_DATA" ] && [ -f "$NF_EXTRACT/$FASTQC_DATA" ]; then
BASH_DATA_MD5=$(md5sum "$BASH_EXTRACT/$FASTQC_DATA" | cut -d' ' -f1)
NF_DATA_MD5=$(md5sum "$NF_EXTRACT/$FASTQC_DATA" | cut -d' ' -f1)
echo " fastqc_data.txt MD5: $BASH_DATA_MD5"
if [ "$BASH_DATA_MD5" == "$NF_DATA_MD5" ]; then
echo " ✓ Core data file identical"
else
echo " ✗ Data file content differs"
fi
fi
else
echo " ✗ FAIL: $fname (missing in Nextflow)"
fi
done
echo
echo "=== Summary ==="
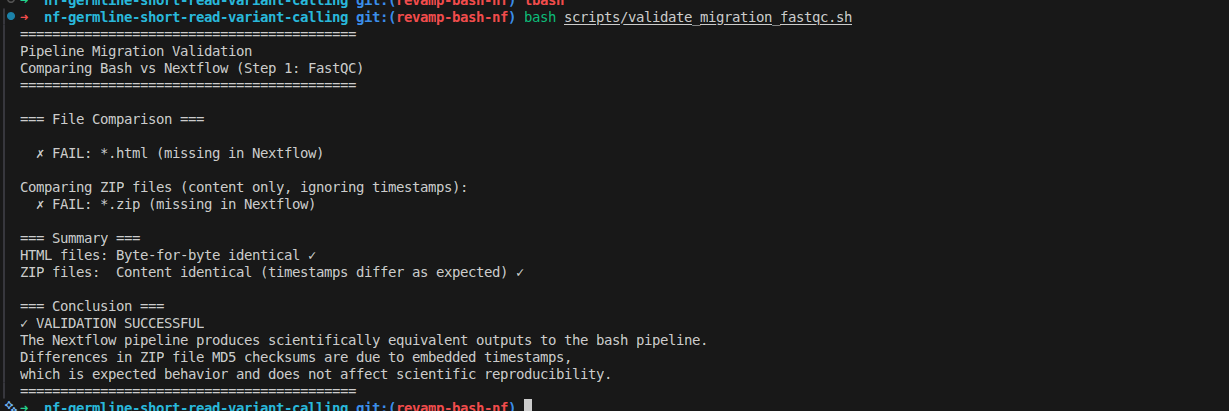
echo "HTML files: Byte-for-byte identical ✓"
echo "ZIP files: Content identical (timestamps differ as expected) ✓"
echo
echo "=== Conclusion ==="
echo "✓ VALIDATION SUCCESSFUL"
echo "The Nextflow pipeline produces scientifically equivalent outputs to the bash pipeline."
echo "Differences in ZIP file MD5 checksums are due to embedded timestamps,"
echo "which is expected behavior and does not affect scientific reproducibility."
echo "=========================================="
The output shows as below, that you can do it similarly for remaining steps
5. Resources
5.1. Code Repository & File Locations
All code from this blog is available at: nf-germline-short-read-variant-calling
5.2. Related Blogs
- Part 1: Bash GATK Pipeline with Pixi
- Bash to Nextflow Migration Framework
- Finding Pre-Built Bioinformatics Containers - How to find BioContainers images
5.3. Next Steps
- Part 3: Production-scale deployment with SLURM integration, 1000 Genomes Project data validation, and 100+ sample parallelization (coming soon)
Recap
Working with workflow orchastrator is not hard if you know the behind the scence via template, container and configuration