Variant Calling (Part 4): Test/Lint Your Nextflow Workflow

In Part 3, we have a working pipeline, but we're missing a key concept: testing and linting workflows. This blog will show you why these practices are essential and how to implement them.
1. Install Extensions and Tools
This tutorial uses Visual Studio Code or Code Server, the most common development environments for Nextflow pipeline development.
1.1 Extension Pack
On the left sidebar, open the Extensions panel and search for extensions.
Check the installed extensions:
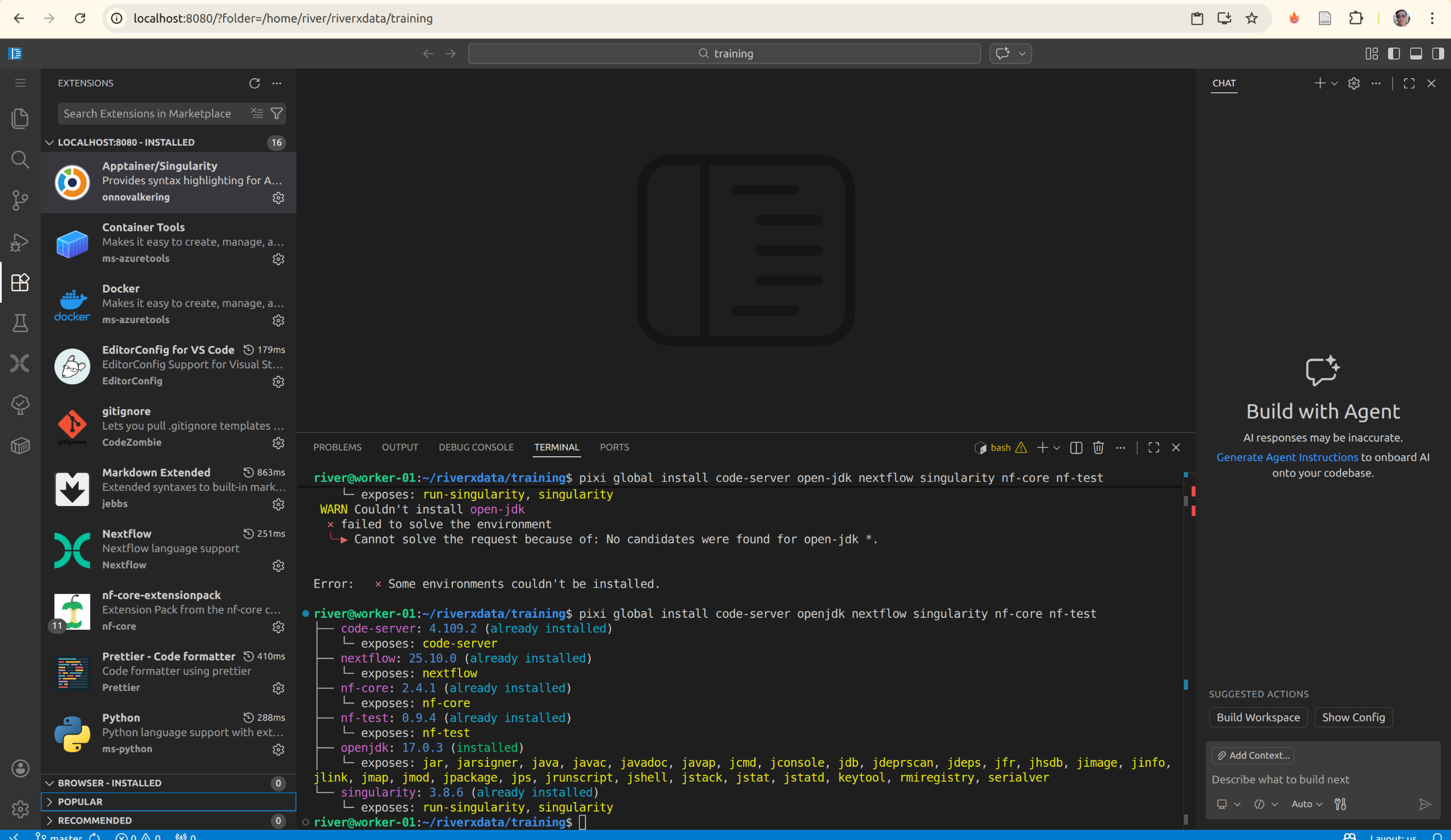
1.2 Nf-core and nf-test
These can be installed via pixi:
pixi add nf-core nf-test python==3.11
2. Getting Started
2.1 Linting
Linting helps your script automatically follow the format that has been previously defined. While you may have the same code logic, it's important to follow consistent indentation, naming conventions, and process organization.
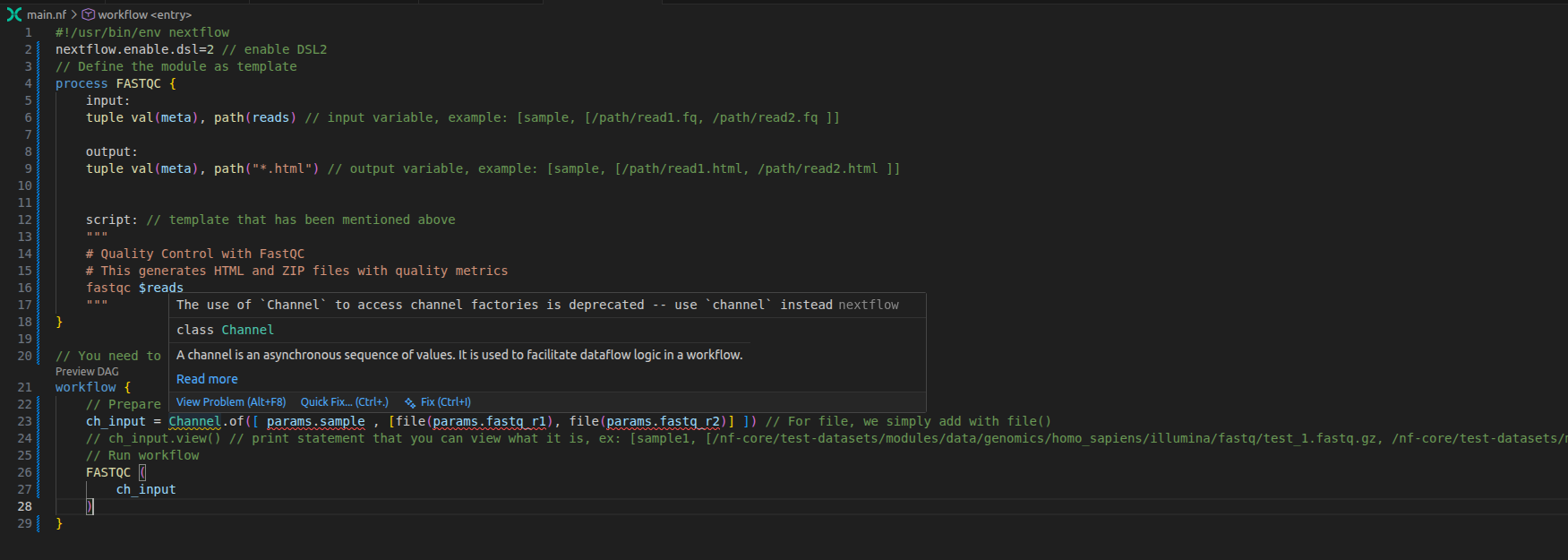
This allows teams to maintain consistent coding conventions, which makes collaboration easier and improves code readability. For example, when you open a simple main.nf script in VS Code, it will highlight violations of the nf-core linting rules.
Manually fix these issues until all warning messages disappear:
Additionally, you can quickly format indentation (spaces and tabs) using the nextflow command:
nextflow lint . -format
- nf-core/tools is a Python tool that provides stricter linting with nf-core conventions: https://github.com/nf-core/tools
- However, this tool enforces certain rules that may not apply to all workflows (e.g., requiring MultiQC, specific file structures). Consider using it based on your use case.
2.2 Testing
In software development, there are several types of tests:
- Unit tests: These tests focus on individual components or functions to ensure they work as expected in isolation. They help catch bugs early and make refactoring safer.
- End-to-end tests: These tests simulate real user scenarios by running the entire workflow from start to finish. They verify that all integrated parts of the system work together correctly.
- Performance tests: These tests measure how the workflow performs under different conditions, such as varying data sizes or system loads, to ensure it meets speed and resource usage requirements.
Applying these testing strategies to your Nextflow workflows helps ensure reliability, maintainability, and reproducibility across different environments.
For bioinformatics applications, I recommend setting up workflows with testing tools to ensure reproducibility across environments. From an engineering perspective, I recommend running small test datasets for new features, and full datasets when releasing to ensure everything works perfectly. You can run tests as follows:
For small dataset tests:
nextflow run main.nf -profile test,docker
For full dataset tests:
nextflow run main.nf -profile test,docker
If both runs complete successfully, the next step is to validate the results. Sometimes a workflow runs successfully but produces unexpected results. From an engineering perspective, we need to ensure that running the same input produces identical results. Using MD5 checksums is a good approach. Check the previous validation blog for similar ideas.
However, manually validating all output files is cumbersome without proper tools or frameworks. Fortunately, nf-test and nft-utils handle this for us.
To set up nf-test, create the files as described below:
2.2.1 Prepare Workflow
You can work with any Nextflow workflow. Here, we'll continue with our nf-germline-short-read-variant-calling repository:
git clone https://github.com/nttg8100/nf-germline-short-read-variant-calling.git -b 0.2.0
cd nf-germline-short-read-variant-calling
2.2.2 Configuration (nf-test.config)
This file tells nf-test where to find tests and which configuration to combine with your workflow:
config {
// location for all nf-test tests
testsDir = "."
// nf-test directory including temporary files for each test
workDir = System.getenv("NFT_WORKDIR") ?: ".nf-test"
// location of an optional nextflow.config file specific for executing tests
configFile = "tests/nextflow.config"
// load the necessary plugins
plugins {
load "nft-utils@0.0.3"
}
}
2.2.3 Test Workflow (default.nf.test)
This file runs the main.nf workflow (the parent workflow in this repository). Comments describe what each section does:
nextflow_pipeline {
name "Test pipeline" // name of workflow
script "../main.nf" // run parent folder workflow
tag "pipeline"
test("Test workflow completion") { // name describing what you're testing; you can define multiple tests here
when {
params { // define your custom parameters here; I copied these from the test profile
outdir = "$outputDir"
input = "$projectDir/assets/samplesheet.csv"
genome = 'sarek_test'
}
}
then {
// stable_name: All files + folders in ${params.outdir}/ with relative paths
def stable_name = getAllFilesFromDir(params.outdir, relative: true, includeDir: true, ignore: ['pipeline_info/*.{html,json,txt}'])
// stable_path: All files in ${params.outdir}/ with stable content
def stable_path = getAllFilesFromDir(params.outdir, ignoreFile: 'tests/.nftignore')
assertAll(
{ assert workflow.success},
{ assert snapshot(
// All stable path names, with relative paths
stable_name,
// All files with stable contents
stable_path
).match() }
)
}
}
}
2.2.4 Ignore Files (.nftignore)
From the previous blog, we know that many tools output files with timestamps. Therefore, we should exclude these when validating to ensure we can reliably compare results. This works similarly to .gitignore:
pipeline_info/*.{html,json,txt,yml}
germline_variant_calling:preprocessing:fastp/*.{html,fastq.gz,json}
germline_variant_calling:preprocessing:gatk_collectmetrics/*.txt
germline_variant_calling*/*.{bcf,vcf,vcf.gz,vcf.gz.tbi}
2.3 Running Test Workflow With Snapshot
nf-test runs similarly to the nextflow command. The --verbose flag shows detailed logs, and --update-snapshot creates the folder structure and MD5 hashes for all output files:
nf-test test tests/default.nf.test --verbose --update-snapshot --profile docker
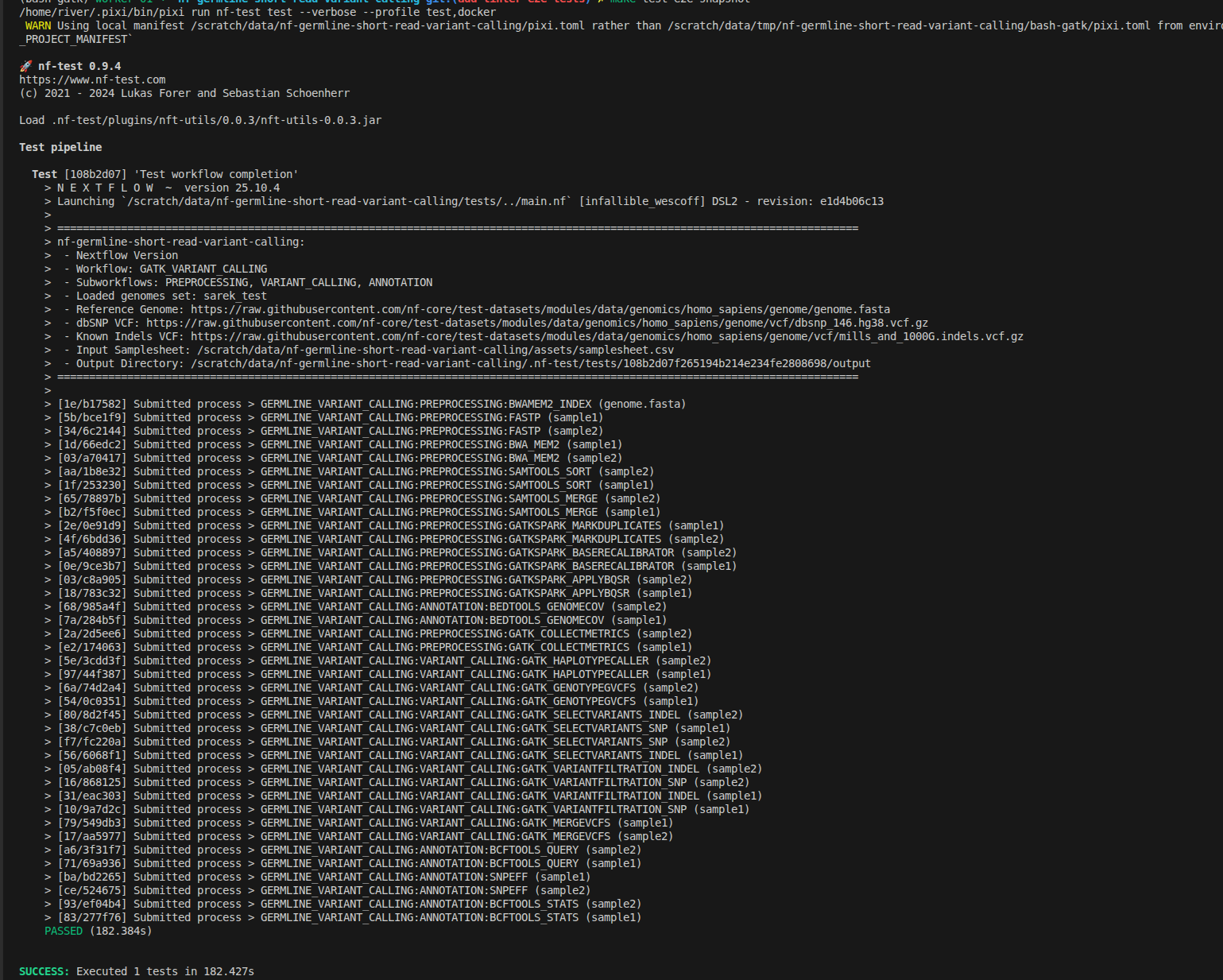
The snapshot file will be automatically created as default.nf.test.snap:
{
"Test workflow completion": {
"content": [
[
"germline_variant_calling:annotation:bcftools_query",
"germline_variant_calling:annotation:bcftools_query/sample1_variants.bed",
"germline_variant_calling:annotation:bcftools_query/sample2_variants.bed",
"germline_variant_calling:annotation:bcftools_query/versions.yml",
"germline_variant_calling:annotation:bcftools_stats",
"germline_variant_calling:annotation:bcftools_stats/sample1_indel_count.txt",
"germline_variant_calling:annotation:bcftools_stats/sample1_snp_count.txt",
"germline_variant_calling:annotation:bcftools_stats/sample1_variant_stats.txt",
"germline_variant_calling:annotation:bcftools_stats/sample2_indel_count.txt",
"germline_variant_calling:annotation:bcftools_stats/sample2_snp_count.txt",
"germline_variant_calling:annotation:bcftools_stats/sample2_variant_stats.txt",
"germline_variant_calling:annotation:bcftools_stats/versions.yml",
"germline_variant_calling:annotation:bedtools_genomecov",
"germline_variant_calling:annotation:bedtools_genomecov/sample1_coverage.bedgraph",
"germline_variant_calling:annotation:bedtools_genomecov/sample2_coverage.bedgraph",
"germline_variant_calling:annotation:bedtools_genomecov/versions.yml",
"germline_variant_calling:annotation:snpeff",
"germline_variant_calling:annotation:snpeff/sample1_annotated.vcf",
"germline_variant_calling:annotation:snpeff/sample1_snpeff.log",
"germline_variant_calling:annotation:snpeff/sample2_annotated.vcf",
"germline_variant_calling:annotation:snpeff/sample2_snpeff.log",
"germline_variant_calling:annotation:snpeff/versions.yml",
"germline_variant_calling:preprocessing:bwa_mem2",
"germline_variant_calling:preprocessing:bwa_mem2/sample1_1_aligned.bam",
"germline_variant_calling:preprocessing:bwa_mem2/sample2_1_aligned.bam",
"germline_variant_calling:preprocessing:bwa_mem2/versions.yml",
"germline_variant_calling:preprocessing:bwamem2_index",
"germline_variant_calling:preprocessing:bwamem2_index/genome.fasta.0123",
"germline_variant_calling:preprocessing:bwamem2_index/genome.fasta.amb",
"germline_variant_calling:preprocessing:bwamem2_index/genome.fasta.ann",
"germline_variant_calling:preprocessing:bwamem2_index/genome.fasta.bwt.2bit.64",
"germline_variant_calling:preprocessing:bwamem2_index/genome.fasta.pac",
"germline_variant_calling:preprocessing:bwamem2_index/versions.yml",
"germline_variant_calling:preprocessing:fastp",
"germline_variant_calling:preprocessing:fastp/sample1_1_fastp.html",
"germline_variant_calling:preprocessing:fastp/sample1_1_fastp.json",
"germline_variant_calling:preprocessing:fastp/sample1_1_trimmed_1.fastq.gz",
"germline_variant_calling:preprocessing:fastp/sample1_1_trimmed_2.fastq.gz",
"germline_variant_calling:preprocessing:fastp/sample2_1_fastp.html",
"germline_variant_calling:preprocessing:fastp/sample2_1_fastp.json",
"germline_variant_calling:preprocessing:fastp/sample2_1_trimmed_1.fastq.gz",
"germline_variant_calling:preprocessing:fastp/sample2_1_trimmed_2.fastq.gz",
"germline_variant_calling:preprocessing:fastp/versions.yml",
"germline_variant_calling:preprocessing:gatk_collectmetrics",
"germline_variant_calling:preprocessing:gatk_collectmetrics/sample1_alignment_summary.txt",
"germline_variant_calling:preprocessing:gatk_collectmetrics/sample2_alignment_summary.txt",
"germline_variant_calling:preprocessing:gatk_collectmetrics/versions.yml",
"germline_variant_calling:preprocessing:gatkspark_applybqsr",
"germline_variant_calling:preprocessing:gatkspark_applybqsr/sample1_recal.bam",
"germline_variant_calling:preprocessing:gatkspark_applybqsr/sample1_recal.bam.bai",
"germline_variant_calling:preprocessing:gatkspark_applybqsr/sample2_recal.bam",
"germline_variant_calling:preprocessing:gatkspark_applybqsr/sample2_recal.bam.bai",
"germline_variant_calling:preprocessing:gatkspark_applybqsr/versions.yml",
"germline_variant_calling:preprocessing:gatkspark_baserecalibrator",
"germline_variant_calling:preprocessing:gatkspark_baserecalibrator/sample1_recal.table",
"germline_variant_calling:preprocessing:gatkspark_baserecalibrator/sample2_recal.table",
"germline_variant_calling:preprocessing:gatkspark_baserecalibrator/versions.yml",
"germline_variant_calling:preprocessing:gatkspark_markduplicates",
"germline_variant_calling:preprocessing:gatkspark_markduplicates/sample1.bam",
"germline_variant_calling:preprocessing:gatkspark_markduplicates/sample1.bam.bai",
"germline_variant_calling:preprocessing:gatkspark_markduplicates/sample2.bam",
"germline_variant_calling:preprocessing:gatkspark_markduplicates/sample2.bam.bai",
"germline_variant_calling:preprocessing:gatkspark_markduplicates/versions.yml",
"germline_variant_calling:preprocessing:samtools_merge",
"germline_variant_calling:preprocessing:samtools_merge/sample1_merged.bam",
"germline_variant_calling:preprocessing:samtools_merge/sample1_merged.bam.bai",
"germline_variant_calling:preprocessing:samtools_merge/sample2_merged.bam",
"germline_variant_calling:preprocessing:samtools_merge/sample2_merged.bam.bai",
"germline_variant_calling:preprocessing:samtools_merge/versions.yml",
"germline_variant_calling:preprocessing:samtools_sort",
"germline_variant_calling:preprocessing:samtools_sort/sample1_sorted.bam",
"germline_variant_calling:preprocessing:samtools_sort/sample2_sorted.bam",
"germline_variant_calling:preprocessing:samtools_sort/versions.yml",
"germline_variant_calling:variant_calling:gatk_genotypegvcfs",
"germline_variant_calling:variant_calling:gatk_genotypegvcfs/sample1_raw.vcf.gz",
"germline_variant_calling:variant_calling:gatk_genotypegvcfs/sample1_raw.vcf.gz.tbi",
"germline_variant_calling:variant_calling:gatk_genotypegvcfs/sample2_raw.vcf.gz",
"germline_variant_calling:variant_calling:gatk_genotypegvcfs/sample2_raw.vcf.gz.tbi",
"germline_variant_calling:variant_calling:gatk_genotypegvcfs/versions.yml",
"germline_variant_calling:variant_calling:gatk_haplotypecaller",
"germline_variant_calling:variant_calling:gatk_haplotypecaller/sample1.g.vcf.gz",
"germline_variant_calling:variant_calling:gatk_haplotypecaller/sample1.g.vcf.gz.tbi",
"germline_variant_calling:variant_calling:gatk_haplotypecaller/sample2.g.vcf.gz",
"germline_variant_calling:variant_calling:gatk_haplotypecaller/sample2.g.vcf.gz.tbi",
"germline_variant_calling:variant_calling:gatk_haplotypecaller/versions.yml",
"germline_variant_calling:variant_calling:gatk_mergevcfs",
"germline_variant_calling:variant_calling:gatk_mergevcfs/sample1_filtered.vcf.gz",
"germline_variant_calling:variant_calling:gatk_mergevcfs/sample1_filtered.vcf.gz.tbi",
"germline_variant_calling:variant_calling:gatk_mergevcfs/sample2_filtered.vcf.gz",
"germline_variant_calling:variant_calling:gatk_mergevcfs/sample2_filtered.vcf.gz.tbi",
"germline_variant_calling:variant_calling:gatk_mergevcfs/versions.yml",
"germline_variant_calling:variant_calling:gatk_selectvariants_indel",
"germline_variant_calling:variant_calling:gatk_selectvariants_indel/sample1_raw_indels.vcf.gz",
"germline_variant_calling:variant_calling:gatk_selectvariants_indel/sample1_raw_indels.vcf.gz.tbi",
"germline_variant_calling:variant_calling:gatk_selectvariants_indel/sample2_raw_indels.vcf.gz",
"germline_variant_calling:variant_calling:gatk_selectvariants_indel/sample2_raw_indels.vcf.gz.tbi",
"germline_variant_calling:variant_calling:gatk_selectvariants_indel/versions.yml",
"germline_variant_calling:variant_calling:gatk_selectvariants_snp",
"germline_variant_calling:variant_calling:gatk_selectvariants_snp/sample1_raw_snps.vcf.gz",
"germline_variant_calling:variant_calling:gatk_selectvariants_snp/sample1_raw_snps.vcf.gz.tbi",
"germline_variant_calling:variant_calling:gatk_selectvariants_snp/sample2_raw_snps.vcf.gz",
"germline_variant_calling:variant_calling:gatk_selectvariants_snp/sample2_raw_snps.vcf.gz.tbi",
"germline_variant_calling:variant_calling:gatk_selectvariants_snp/versions.yml",
"germline_variant_calling:variant_calling:gatk_variantfiltration_indel",
"germline_variant_calling:variant_calling:gatk_variantfiltration_indel/sample1_filtered_indels.vcf.gz",
"germline_variant_calling:variant_calling:gatk_variantfiltration_indel/sample1_filtered_indels.vcf.gz.tbi",
"germline_variant_calling:variant_calling:gatk_variantfiltration_indel/sample2_filtered_indels.vcf.gz",
"germline_variant_calling:variant_calling:gatk_variantfiltration_indel/sample2_filtered_indels.vcf.gz.tbi",
"germline_variant_calling:variant_calling:gatk_variantfiltration_indel/versions.yml",
"germline_variant_calling:variant_calling:gatk_variantfiltration_snp",
"germline_variant_calling:variant_calling:gatk_variantfiltration_snp/sample1_filtered_snps.vcf.gz",
"germline_variant_calling:variant_calling:gatk_variantfiltration_snp/sample1_filtered_snps.vcf.gz.tbi",
"germline_variant_calling:variant_calling:gatk_variantfiltration_snp/sample2_filtered_snps.vcf.gz",
"germline_variant_calling:variant_calling:gatk_variantfiltration_snp/sample2_filtered_snps.vcf.gz.tbi",
"germline_variant_calling:variant_calling:gatk_variantfiltration_snp/versions.yml",
"pipeline_info"
],
[
"sample1_variants.bed:md5,d41d8cd98f00b204e9800998ecf8427e",
"sample2_variants.bed:md5,d41d8cd98f00b204e9800998ecf8427e",
"versions.yml:md5,249943b1d5bd2ce57e04bea4d2dd8abf",
"sample1_indel_count.txt:md5,897316929176464ebc9ad085f31e7284",
"sample1_snp_count.txt:md5,897316929176464ebc9ad085f31e7284",
"sample1_variant_stats.txt:md5,e8e84ea2acff4d7b5548b7ebaf2b05a4",
"sample2_indel_count.txt:md5,897316929176464ebc9ad085f31e7284",
"sample2_snp_count.txt:md5,897316929176464ebc9ad085f31e7284",
"sample2_variant_stats.txt:md5,2301b862009bbf518a5f94f19103b5de",
"versions.yml:md5,ff6ad194e0ec804b72bc83d8bf8cf019",
"sample1_coverage.bedgraph:md5,d41d8cd98f00b204e9800998ecf8427e",
"sample2_coverage.bedgraph:md5,d41d8cd98f00b204e9800998ecf8427e",
"versions.yml:md5,6d58041eb279541e3a138c60826cefbb",
"sample1_snpeff.log:md5,7137fc9f7d044314bea421ba06ca0f18",
"sample2_snpeff.log:md5,7137fc9f7d044314bea421ba06ca0f18",
"versions.yml:md5,1d4d4e229cfa017c03d08c73b5674b98",
"sample1_1_aligned.bam:md5,90cf54bb11be1393f24791f2bafb81ba",
"sample2_1_aligned.bam:md5,ecc75033f0c0c9a88497799323cd12af",
"versions.yml:md5,f1355436d7ddd31b50858fe62e8adde4",
"genome.fasta.0123:md5,d73300d44f733bcdb7c988fc3ff3e3e9",
"genome.fasta.amb:md5,1891c1de381b3a96d4e72f590fde20c1",
"genome.fasta.ann:md5,2df4aa2d7580639fa0fcdbcad5e2e969",
"genome.fasta.bwt.2bit.64:md5,cd4bdf496eab05228a50c45ee43c1ed0",
"genome.fasta.pac:md5,8569fbdb2c98c6fb16dfa73d8eacb070",
"versions.yml:md5,2a4b9defcea58647c0b5b1cc8b2627f2",
"versions.yml:md5,6c7466de4bab59e8df203424eb253fd6",
"versions.yml:md5,5fa3be89c5df1c030acae287599419dd",
"sample1_recal.bam:md5,92a4e508f76d8dfe7a7149ad8861feb3",
"sample1_recal.bam.bai:md5,0d76977b2e36046cc176112776c5fa4e",
"sample2_recal.bam:md5,4a4de50378f16f4ff1345c61c50df2fb",
"sample2_recal.bam.bai:md5,0d76977b2e36046cc176112776c5fa4e",
"versions.yml:md5,04197383659004cbbc3bcd59260d5bc0",
"sample1_recal.table:md5,c38e5c25e24ced2c98c908cf89328ecf",
"sample2_recal.table:md5,c38e5c25e24ced2c98c908cf89328ecf",
"versions.yml:md5,c935190eb4847737950a118a8ef8ef53",
"sample1.bam:md5,92a4e508f76d8dfe7a7149ad8861feb3",
"sample1.bam.bai:md5,0d76977b2e36046cc176112776c5fa4e",
"sample2.bam:md5,4a4de50378f16f4ff1345c61c50df2fb",
"sample2.bam.bai:md5,0d76977b2e36046cc176112776c5fa4e",
"versions.yml:md5,921b80662856f7496e4d181748b284ab",
"sample1_merged.bam:md5,663f5532903b2fb669dc39fd58bfd387",
"sample1_merged.bam.bai:md5,0d76977b2e36046cc176112776c5fa4e",
"sample2_merged.bam:md5,137440788103c2ce509a89169d8e1a2c",
"sample2_merged.bam.bai:md5,0d76977b2e36046cc176112776c5fa4e",
"versions.yml:md5,df0d93935f80356c470858f91ee34f47",
"sample1_sorted.bam:md5,663f5532903b2fb669dc39fd58bfd387",
"sample2_sorted.bam:md5,137440788103c2ce509a89169d8e1a2c",
"versions.yml:md5,f30c0e213665fdb93c19cde77cd1b967",
"versions.yml:md5,28ef44a18a3310ca11717b590731eadc",
"versions.yml:md5,1a07831fb1f0ee9f9248c68736c30695",
"versions.yml:md5,8a62c99aa5d272e89ff0935055091745",
"versions.yml:md5,d6c0b7ff8e09b3c996ef151bd5eb546a",
"versions.yml:md5,cbda79c8ab968bd76b73fd7c739dcca3",
"versions.yml:md5,dc9ae42e4bea47b9da0c0f9a905c0684",
"versions.yml:md5,d3949b20e639f1016e9745b2a1126960"
]
],
"timestamp": "2026-03-04T23:25:53.360079629",
"meta": {
"nf-test": "0.9.4",
"nextflow": "25.10.4"
}
}
}
2.4 Running Tests With Existing Snapshots
Now that you've completed the initial testing, you don't need to update snapshots again. You can simply run the test to ensure it matches the existing snapshot:
nf-test test tests/default.nf.test --verbose --profile docker
2.5 Optional (Manual Testing)
The workflow should run successfully, but you may want to add tests that validate file contents to ensure the results are meaningful. For example, you could simulate a dataset with specific variants in known regions and allele frequencies. After completing the test, you can write a Python script to validate that the workflow correctly calls these variants:
python validate_germline_variant_calling.py --expect-variant truth.vcf --pipeline-variant query.vcf
3. GitHub Actions With Linting
3.1 Makefile
In my previous blog, I introduced the Makefile, which sets up a consistent environment for both local development and GitHub Actions. Using GitHub Actions' pre-defined actions can be harder to debug in my experience. Instead, I use a Makefile to quickly run tests locally rather than running end-to-end in GitHub Actions workflow files.
Get started with CI/CD in bioinformatics: CI/CD in bioinformatics
I create a single Makefile with test and lint targets bound together:
.PHONY: test-e2e clean
${HOME}/.pixi/bin/pixi:
curl -sSL https://pixi.sh/install.sh | sh
test-e2e: ${HOME}/.pixi/bin/pixi
${HOME}/.pixi/bin/pixi run nextflow run main.nf -profile docker,test -resume
test-e2e-snapshot: ${HOME}/.pixi/bin/pixi
${HOME}/.pixi/bin/pixi run nf-test test --verbose --profile test,docker
test-e2e-update-snapshot: ${HOME}/.pixi/bin/pixi
${HOME}/.pixi/bin/pixi run nf-test test tests/default.nf.test --verbose --update-snapshot --profile test,docker
lint: ${HOME}/.pixi/bin/pixi
${HOME}/.pixi/bin/pixi run nextflow lint . -format
clean:
rm -rf work
3.2 GitHub Workflow
The successfull run of Gihub Actions can be found on the PR https://github.com/nttg8100/nf-germline-short-read-variant-calling/pull/7:
- Linter: https://github.com/nttg8100/nf-germline-short-read-variant-calling/actions/runs/22679273108
- Test: https://github.com/nttg8100/nf-germline-short-read-variant-calling/actions/runs/22679273035
3.2.1 Linting
Create the file .github/workflows/linting.yaml. This workflow:
- Clones your repository and runs the lint command to ensure code is properly formatted before pushing to GitHub
- Triggers on push and pull requests to the
mainbranch
name: Linting
on:
push:
branches: [ main ]
pull_request:
branches: [ main ]
jobs:
clone:
runs-on: ubuntu-latest
steps:
- name: Checkout repository
uses: actions/checkout@v4
- name: Running linting
run: |
make lint
3.2.2 Testing
Create the file .github/workflows/e2e.yml. This workflow:
- Clones your repository and runs the pipeline with testing output snapshots
- Triggers on push and pull requests to the
mainbranch
name: Testing e2e
on:
push:
branches: [ main ]
pull_request:
branches: [ main ]
jobs:
clone:
runs-on: ubuntu-latest
steps:
- name: Checkout repository
uses: actions/checkout@v4
- name: Running testing
run: |
make test-e2e-snapshot
Recap
These core testing and linting principles can be applied not only to Nextflow but to any workflow to achieve best practices.