Variant Calling (Part 1): Building a Reproducible GATK Variant Calling Bash Workflow with Pixi

This blog is designed as a practical starting point for building bioinformatics workflows focused on germline variant calling. You'll begin with a straightforward, standard approach using bash and reproducible environments. In future posts, we'll explore how to transition to best-practice workflow management with Nextflow, allowing for further optimization, customization, and integration of additional tools to enhance workflow quality.
This tutorial has been released under Github repository: https://github.com/nttg8100/nf-germline-short-read-variant-calling/tree/0.1.0
Series Overview
This is Part 1 of a two-part series on modernizing bioinformatics workflows:
-
Part 1 (This Post): Build simply bash script workflow, ensure the reproducible environments
-
Part 2 (Coming Soon): Transforming to Nextflow, ensure the consistency on the environments and results
Prerequisites & Related Topics
Before diving into this blog, readers should be familiar with:
- Pixi - New conda era - Essential for understanding the package management setup
- Containers on HPC: From Docker to Singularity and Apptainer - Container fundamentals for reproducibility
- The Evolution of Version Control - Git's Role in Reproducible Bioinformatics (Part 1) - Version control basics
1. The GATK Variant Calling Workflow
1.1. Understanding the NGS101 Workflow
The NGS101 GATK tutorial offers a clear introduction to the fundamentals of germline variant calling workflows. In this section, we’ll break down the 10 essential steps from that tutorial to help you understand the basic structure of a variant calling pipeline.
Germline variant calling is a core part of DNA sequencing analysis using whole genome data. DNA, our genetic material, consists of 23 pairs of chromosomes and over 3 billion base pairs. While all humans share a similar DNA structure, the exact sequence of nucleotides (A, T, C, G) varies between individuals—these differences are called variants.
The Human Genome Project established a reference genome, which serves as a template for comparing individual genomes. Variants are the small differences between an individual’s DNA and this reference. Most variants are benign and simply contribute to our uniqueness.
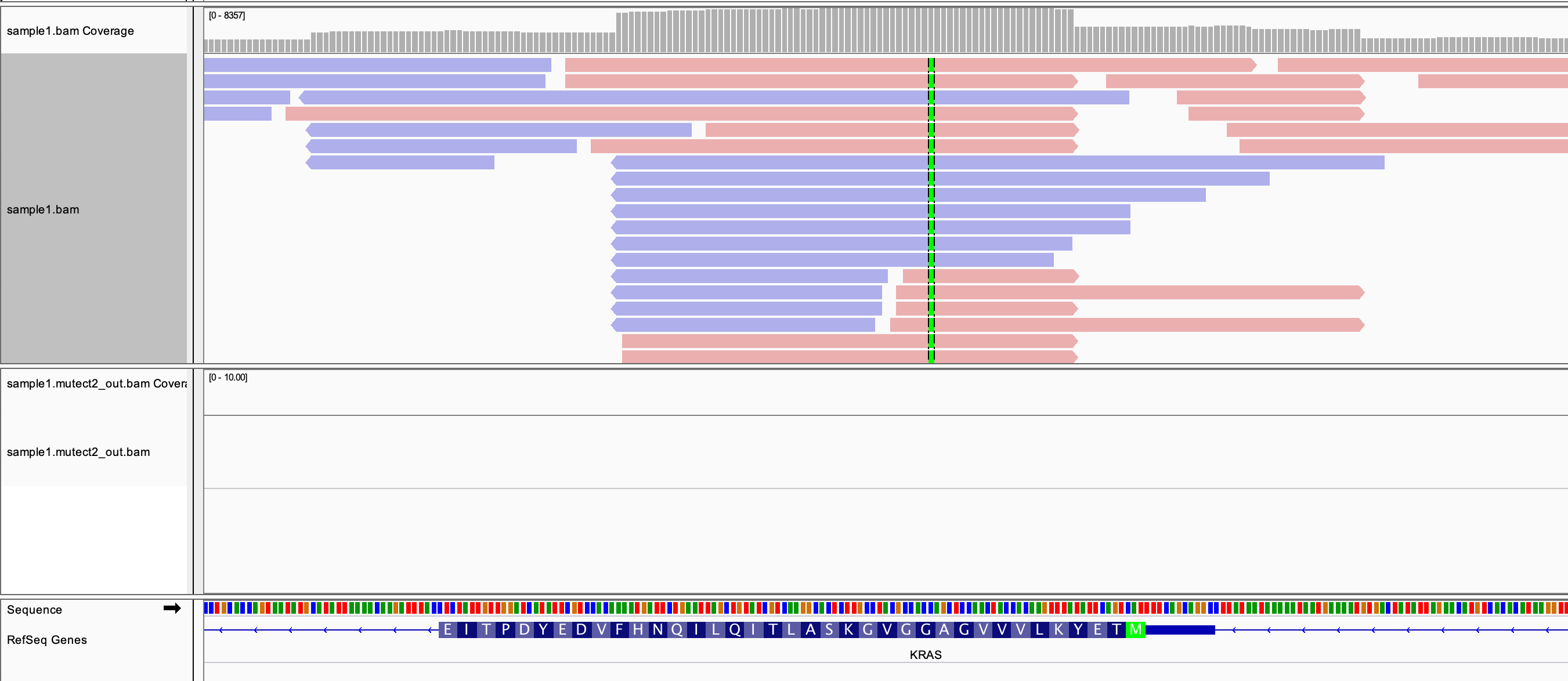
The figure below illustrates how paired-end sequencing reads (shown in red and blue) are aligned to the reference genome. When a read differs from the reference at a specific position (highlighted in green), it suggests the presence of a variant. If multiple reads support the same difference, the evidence for a true variant increases.
To confidently identify variants, sequencing reads (short DNA fragments) must be accurately aligned to the reference genome. Germline variants are inherited and typically appear in about 40–50% of reads (heterozygous, inherited from one parent) or 100% (homozygous, inherited from both parents). In contrast, somatic mutations arise in non-inherited cells (e.g., cancer) and often require comparing tumor and normal samples to distinguish true somatic variants from inherited ones.
By following these foundational steps, you can build a robust workflow for germline variant calling and understand the biological context behind each stage.
 Reference:
Reference: 1.2 Data
There are many publicly available datasets for variant calling workflow development. For this tutorial, I use the small test dataset provided by nf-core/sarek, which is designed for rapid pipeline prototyping.
A key step in workflow development is working with a manageable, representative subset of data. How do projects like sarek create these small datasets? Typically, they extract a subset of raw reads that align to a specific genomic region. For example, you can align the original FASTQ files to the reference genome using a simple bash script, then extract only the reads mapped to a region such as chr22:100,000-200,000. Similarly, you can subset the reference genome to just chr22:1-250,000. This approach dramatically reduces input data size and speeds up pipeline testing and iteration. Once your workflow is validated on the small dataset, you can scale up to the full dataset for comprehensive analysis.
1.3 Steps Explaination
1.3.1 Quality Control Raw Reads (FastQC)
- Assess read quality metrics
- Identify potential sequencing issues
- Generate HTML reports
# download raw small data
wget https://raw.githubusercontent.com/nf-core/test-datasets/modules/data/genomics/homo_sapiens/illumina/fastq/test_1.fastq.gz
# view file
zcat test_1.fastq.gz|head
Understanding FASTQ File Structure
@normal#21#998579#1/1
CCTTCTCCCTGCTGGGGTTGCTTGTCAGTAGCGGGCAAGGTAGGAGTGTGGCGCTTTATTGCATTTACTTTCCCTCCCCCTTCCCCCCGGCCAAGAGAGG
+
102302000331;3333;23133320233330*33/233333333333333/313232333/3;3;3/333000;11/00;;01//103*1032323233
@normal#21#998572#2/1
CTCCTCTCCTTCTACCTGCTGGGGTTGCTTGTCAGTAGCGGGCAAGGTCGGAGTGTTGCGCTTTATTGCATTTACTTTCCCTCCCCCTTCCACCCGGCCA
+
/201302/0330;30033133333;230;333033333033300333203333333;*03133;223300;2;3/33211/210001;2/13/0133//3
@normal#21#998620#3/1
AGGAGTGGGCGCTTTATTGCATTTACTTTCCGTCCCCCTTCCACCCGGCCAAGAGAGGCAAAGAGGGAGGCCAGGAAGAATGAAATGAAAGAACTAAA
A FASTQ file is the standard format for storing raw sequencing reads from NGS platforms. Each read is represented by four lines:
- Sequence Identifier (starts with
@): Contains information about the read (e.g., instrument, run, coordinates). - Nucleotide Sequence: The actual DNA bases (A, T, C, G, or N).
- Separator Line (starts with
+): May repeat the identifier or be a simple plus sign. - Quality Scores: ASCII-encoded Phred quality scores for each base in the sequence.
Example:
@normal#21#998579#1/1
CCTTCTCCCTGCTGGGGTTGCTTGTCAGTAGCGGGCAAGGTAGGAGTGTGGCGCTTTATTGCATTTACTTTCCCTCCCCCTTCCCCCCGGCCAAGAGAGG
+
102302000331;3333;23133320233330*33/233333333333333/313232333/3;3;3/333000;11/00;;01//103*1032323233
- The first line is the read name.
- The second line is the sequence.
- The third line is a separator.
- The fourth line encodes the quality of each base (same length as the sequence).
This structure repeats for every read in the file. We usually have tool to trim the reads that has the low quality reads. Next step, if not, it may not align well to the reference genome or cause the miss variant calling. Low quality reads are the reference genome.
1.3.2 Trim Reads (Trimgalore)
- It is simply to trim the reads with low quality.
- With trimgalore, it reruns the FastQC to check the trimmed reads data to ensure they are trimmed correctly, not too much low quality reads are removed.
1.3.3 Aligned to the reference genome
Download and see how the reference genome look likes
# Download reference genome
wget https://raw.githubusercontent.com/nf-core/test-datasets/modules/data/genomics/homo_sapiens/genome/genome.fasta
head genome.fasta
You can see the blow is the first 10 lines, it just shows the order of the reference genome in A,T,C,G. However, comparing manually between the
read sequence verus the reference genome is not a good idea. Here, we use the bwa a tool that can help to do it faster. I highly recommend to watch videos
by Ben Langmead where he explain the magic of the algorithm to solve this challanges: https://www.youtube.com/watch?v=6BJbEWyO_N0&list=PL2mpR0RYFQsADmYpW2YWBrXJZ_6EL_3nu&index=4
>chr22
ACTCAAGATAATGATGAGTAAAGAATATATTTCTAACAACAAAAAGGAAATTTGATAGTA
TTTCTAAAGACAAAAAGGAAATTTGTATTCACATTCAGTTAGTCATTCCACCAGAATGAC
TTCATCACACAATATTTTGTGACAAGAACCTGAACAGCCTCATGTTTTACAATATTCTTT
TCATCTTTTATTATATGCACCAAAATTTTCTTTTTTAAATTTTCTTGAACCTCTAAATCT
ACTTTAAAAATTTACCTGATACACTTTTTAAATGGACAAATGCTGAAGGTAGCTGTGTAT
ACAAATGTGACTAGAAGGAAAAAGATGATGTAGAAATACAATAACTCCTTGAGTTGATCA
TTCTGATTGGCATTTATAGAGTAGAAATGTTTTGTAATTACAGAGGAAAAAAGATGGCCT
TTCCTTCAACAGTTATGAGCCGTCAGAATTTTCAAAAATATTGCATTTTGACAATGTAGT
TTCTAGTTTGACAATGATATATTTATCTTCAAAACCAGGAAAATGTAGATAAGGATTTGG
We should index the reference genome once, then using it instead of rerunning for again without changing the reference genome
# simple command, however, to install bwa, later we use pixi
bwa index genome.fasta
1.3.4 Sort and Index Aligned Reads (Samtools)
After alignment, the reads are sorted by their genomic coordinates using Samtools. Sorting is essential for downstream analysis, especially for variant calling and visualization, as many tools expect sorted BAM files. Indexing creates a .bai file, which acts like a table of contents, allowing rapid access to specific regions in the BAM file—similar to quickly finding a chapter in a book.
1.3.5 Mark Duplicates (GATK MarkDuplicates)
GATK's MarkDuplicates identifies and marks PCR or optical duplicate reads—those with identical sequences and alignment positions. Since DNA is fragmented into short reads, true duplicates are rare; most duplicates arise from technical artifacts. Removing or marking these duplicates prevents them from inflating variant support, ensuring more accurate variant calling. For example, if 14% of reads support a variant but 10% are duplicates, only 4% represent true evidence.
1.3.6 Base Quality Score Recalibration (GATK BaseRecalibrator)
This step models and corrects systematic errors in base quality scores assigned by the sequencer. Using known variant sites, GATK BaseRecalibrator generates a recalibration table that helps adjust quality scores, improving the accuracy of downstream variant calls.
# Download one db, we still have indel database
wget https://raw.githubusercontent.com/nf-core/test-datasets/modules/data/genomics/homo_sapiens/genome/vcf/dbsnp_146.hg38.vcf.gz
# Check first few line, it shows the variant at specific position
zcat dbsnp_146.hg38.vcf.gz|grep -v "#"|head|column -t
chr22 66 rs568512106 A G . PASS RS=568512106;RSPOS=16570065;dbSNPBuildID=142;SSR=0;SAO=0;VP=0x050000000005040024000100;WGT=1;VC=SNV;ASP;VLD;KGPhase3;CAF=0.9994,0.000599;COMMON=0
chr22 67 rs371364157 A G . PASS RS=371364157;RSPOS=16570066;dbSNPBuildID=138;SSR=0;SAO=0;VP=0x050000000005000002000100;WGT=1;VC=SNV;ASP
chr22 68 rs536054182 A C . PASS RS=536054182;RSPOS=16570067;dbSNPBuildID=142;SSR=0;SAO=0;VP=0x050000000005040024000100;WGT=1;VC=SNV;ASP;VLD;KGPhase3;CAF=0.9994,0.000599;COMMON=0
chr22 91 rs149338188 ACATT A . PASS RS=149338188;RSPOS=16570091;dbSNPBuildID=134;SSR=0;SAO=0;VP=0x05000000000515003e000200;WGT=1;VC=DIV;ASP;VLD;G5;KGPhase1;KGPhase3;CAF=0.8083,0.1917;COMMON=1
chr22 91 rs75245162 ACATT A . PASS RS=75245162;RSPOS=16570093;dbSNPBuildID=131;SSR=0;SAO=0;VP=0x050000000005000002000200;WGT=1;VC=DIV;ASP;CAF=0.8083,0.1917;COMMON=1
chr22 96 rs150045226 C T . PASS RS=150045226;RSPOS=16570095;dbSNPBuildID=134;SSR=0;SAO=0;VP=0x050000000005000002000100;WGT=1;VC=SNV;ASP
chr22 103 rs563344083 TC T . PASS RS=563344083;RSPOS=16570103;dbSNPBuildID=142;SSR=0;SAO=0;VP=0x050000000005040026000200;WGT=1;VC=DIV;ASP;VLD;KGPhase3;CAF=0.997,0.002995;COMMON=1
chr22 140 rs370473918 T C . PASS RS=370473918;RSPOS=16570139;dbSNPBuildID=138;SSR=0;SAO=0;VP=0x050000000005000002000100;WGT=1;VC=SNV;ASP
chr22 177 rs553984036 C A . PASS RS=553984036;RSPOS=16570176;dbSNPBuildID=142;SSR=0;SAO=0;VP=0x050000000005040024000100;WGT=1;VC=SNV;ASP;VLD;KGPhase3;CAF=0.9996,0.0003994;COMMON=0
chr22 208 rs565829750 T C . PASS RS=565829750;RSPOS=16570207;dbSNPBuildID=142;SSR=0;SAO=0;VP=0x050000000005000024000100;WGT=1;VC=SNV;ASP;KGPhase3;CAF=0.9998,0.0001997;COMMON=0
1.3.7 Apply BQSR (GATK ApplyBQSR)
The recalibration table is applied to the BAM file, updating base quality scores. This produces an analysis-ready BAM file with more reliable quality metrics, which is crucial for confident variant detection.
1.3.8 Quality Assessment
Collect the alignment summary metrics
1.3.9 Variant Calling (GATK HaplotypeCaller)
GATK HaplotypeCaller analyzes the recalibrated BAM file to identify SNPs and indels. It performs local assembly of haplotypes and outputs variants in VCF or GVCF format, providing the foundation for further genotyping and filtering.
1.3.10 Genotype (GATK GenotypeGVCFs)
Genotyping is the process of determining the genetic variants (alleles) present at specific positions in a sample's DNA. GATK GenotypeGVCFs combines variant calls from multiple samples (in GVCF format) to produce a final set of genotypes, enabling accurate identification of variants across a cohort.
2. Install Tools
2.1. Why Pixi Over Conda?
Pixi offers several advantages for bioinformatics workflows:
- Fast: 10x faster than conda for environment resolution
- Reproducible: Lock files ensure identical environments
- Simple: Single
pixi.tomlfile defines everything - Cross-platform: Works on Linux, macOS, and Windows
Follow this blog for deeper understand of how to use pixi more effectively and more efficiently:
- Pixi - New conda era** - Understanding modern package management with Pixi
2.2. Creating the Project
Create a new directory and initialize the Pixi project:
mkdir bash-gatk
cd bash-gatk
# install Pixi if not already installed
curl -fsSL https://pixi.sh/install.sh | bash
pixi init
# activate env that is similar to conda
pixi shell
Create a pixi.toml file, you can use command line or code editor to edit this file:
[workspace]
name = "bash-gatk"
version = "0.1.0"
description = "GATK variant calling workflow - from bash to Nextflow transformation"
channels = ["conda-forge", "bioconda"]
platforms = ["linux-64"]
[dependencies]
To install a tool, you can simple run as below command
# add bwa tools, better with hard version, pixi add lock file while adding hard version that we can quickly check version on the config file
pixi add bwa==0.7.17
# show where you tool is installed
which bwa
You can gradually install tool by tool, step by step. Here is all what we need for the whole standard workflow concept
[workspace]
name = "bash-gatk"
version = "0.1.0"
description = "GATK variant calling workflow - from bash to Nextflow transformation"
channels = ["conda-forge", "bioconda"]
platforms = ["linux-64"]
[dependencies]
bwa = "==0.7.17"
samtools = "==1.16.1"
gatk4 = "==4.6.2.0"
fastqc = "==0.12.1"
curl = "*"
trim-galore = "==0.6.11"
r-base = "==4.4.0"
3. Prepare Inputs And References
It is simply download the input data that runs once to reuse. You do not need to rerun again
#!/bin/bash
#
# Download test data for GATK variant calling pipeline
# This script downloads reference genome, known sites, and test FASTQ files from nf-core
#
set -euo pipefail
echo "=========================================="
echo "Downloading GATK Pipeline Test Data"
echo "=========================================="
echo
# Create directories
echo "Creating directories..."
mkdir -p data reference
echo "✓ Directories created"
echo
# Download reference genome
echo "Downloading reference genome..."
cd reference
curl -L -o genome.fasta \
https://raw.githubusercontent.com/nf-core/test-datasets/modules/data/genomics/homo_sapiens/genome/genome.fasta
echo "✓ genome.fasta downloaded"
curl -L -o genome.fasta.fai \
https://raw.githubusercontent.com/nf-core/test-datasets/modules/data/genomics/homo_sapiens/genome/genome.fasta.fai
echo "✓ genome.fasta.fai downloaded"
curl -L -o genome.dict \
https://raw.githubusercontent.com/nf-core/test-datasets/modules/data/genomics/homo_sapiens/genome/genome.dict
echo "✓ genome.dict downloaded"
# Download known sites for BQSR
echo ""
echo "Downloading known sites VCF files..."
curl -L -o dbsnp_146.hg38.vcf.gz \
https://raw.githubusercontent.com/nf-core/test-datasets/modules/data/genomics/homo_sapiens/genome/vcf/dbsnp_146.hg38.vcf.gz
echo "✓ dbsnp_146.hg38.vcf.gz downloaded"
curl -L -o dbsnp_146.hg38.vcf.gz.tbi \
https://raw.githubusercontent.com/nf-core/test-datasets/modules/data/genomics/homo_sapiens/genome/vcf/dbsnp_146.hg38.vcf.gz.tbi
echo "✓ dbsnp_146.hg38.vcf.gz.tbi downloaded"
curl -L -o mills_and_1000G.indels.vcf.gz \
https://raw.githubusercontent.com/nf-core/test-datasets/modules/data/genomics/homo_sapiens/genome/vcf/mills_and_1000G.indels.vcf.gz
echo "✓ mills_and_1000G.indels.vcf.gz downloaded"
curl -L -o mills_and_1000G.indels.vcf.gz.tbi \
https://raw.githubusercontent.com/nf-core/test-datasets/modules/data/genomics/homo_sapiens/genome/vcf/mills_and_1000G.indels.vcf.gz.tbi
echo "✓ mills_and_1000G.indels.vcf.gz.tbi downloaded"
cd ..
# Index the reference genome
echo ""
echo "Creating BWA index for reference genome..."
echo "This may take a few moments..."
bwa index reference/genome.fasta
echo "✓ BWA index created"
# Download test FASTQ files
echo ""
echo "Downloading test FASTQ files..."
cd data
curl -L -o sample1_R1.fastq.gz \
https://raw.githubusercontent.com/nf-core/test-datasets/modules/data/genomics/homo_sapiens/illumina/fastq/test_1.fastq.gz
echo "✓ test_1.fastq.gz downloaded"
curl -L -o sample1_R2.fastq.gz \
https://raw.githubusercontent.com/nf-core/test-datasets/modules/data/genomics/homo_sapiens/illumina/fastq/test_2.fastq.gz
echo "✓ test_2.fastq.gz downloaded"
cd ..
# Summary
echo ""
echo "=========================================="
echo "Data Download Completed Successfully!"
echo "=========================================="
echo ""
echo "Downloaded files:"
echo "Reference genome:"
ls -lh reference/genome.fasta* reference/genome.dict 2>/dev/null || true
echo ""
echo "BWA indices:"
ls -lh reference/genome.fasta.{amb,ann,bwt,pac,sa} 2>/dev/null || true
echo ""
echo "Known sites VCFs:"
ls -lh reference/*.vcf.gz* 2>/dev/null || true
echo ""
echo "Test FASTQ files:"
ls -lh data/*.fastq.gz 2>/dev/null || true
echo ""
echo "=========================================="
echo "You can now run the pipeline with: Nextflow workflow"
echo " cd nextflow-gatk; pixi run nextflow run main.nf -profile singularity --input samplesheet.csv"
echo "You can now run the pipeline with: Bash workflow"
echo " cd nextflow-gatk; pixi run bash gatk_pipeline.sh"
echo "=========================================="
4. Run End to End
4.1. Run pipeline
The workflow should have this structure
├── bash-gatk
│ ├── gatk_pipeline.sh
│ ├── pixi.lock
│ └── pixi.toml
├── README.md
└── scripts
├── download_data.sh
Now you can run to download the data first, then run the variant calling pipeline later
# Activate env on this folder
pixi shell --manifest-path bash-gatk
# Run the download script (uses Pixi environment for BWA indexing)
bash scripts/download_data.sh
Check the prepared inputs data, where data folder reference folder are correctly configured
├── bash-gatk
│ ├── gatk_pipeline.sh
│ ├── pixi.lock
│ └── pixi.toml
├── data
│ ├── sample1_R1.fastq.gz
│ └── sample1_R2.fastq.gz
├── README.md
├── reference
│ ├── dbsnp_146.hg38.vcf.gz
│ ├── dbsnp_146.hg38.vcf.gz.tbi
│ ├── genome.dict
│ ├── genome.fasta
│ ├── genome.fasta.amb
│ ├── genome.fasta.ann
│ ├── genome.fasta.bwt
│ ├── genome.fasta.fai
│ ├── genome.fasta.pac
│ ├── genome.fasta.sa
│ ├── mills_and_1000G.indels.vcf.gz
│ └── mills_and_1000G.indels.vcf.gz.tbi
└── scripts
├── download_data.sh
Run the bash script workflow where he workflow bash-gatk/gatk_pipeline.sh:
#!/bin/bash
#
# GATK Variant Calling Pipeline - Complete Best Practices (10 Steps)
# Based on NGS101 tutorial: https://ngs101.com/how-to-analyze-whole-genome-sequencing-data-for-absolute-beginners-part-1-from-raw-reads-to-high-quality-variants-using-gatk/
# Adapted to use nf-core test data with hard filtering (Option 2)
#
# This pipeline implements the complete GATK workflow with:
# - Adapter trimming and quality filtering (Trim Galore)
# - Read alignment with BWA-MEM
# - Duplicate marking (GATK MarkDuplicates)
# - Base quality score recalibration (BQSR)
# - Alignment quality metrics (GATK CollectMetrics)
# - Variant calling with HaplotypeCaller (GVCF mode)
set -euo pipefail
# Configuration
SAMPLE="${1:-sample1}" # Accept sample name as first argument, default to sample1
THREADS=2
DATA_DIR="data"
REF_DIR="reference"
RESULTS_DIR="results"
# Reference files
REFERENCE="${REF_DIR}/genome.fasta"
DBSNP="${REF_DIR}/dbsnp_146.hg38.vcf.gz"
KNOWN_INDELS="${REF_DIR}/mills_and_1000G.indels.vcf.gz"
# Input FASTQ files
FASTQ_R1="${DATA_DIR}/${SAMPLE}_R1.fastq.gz"
FASTQ_R2="${DATA_DIR}/${SAMPLE}_R2.fastq.gz"
# Output directories
QC_DIR="${RESULTS_DIR}/qc/${SAMPLE}"
TRIMMED_DIR="${RESULTS_DIR}/trimmed/${SAMPLE}"
ALIGNED_DIR="${RESULTS_DIR}/aligned/${SAMPLE}"
VAR_DIR="${RESULTS_DIR}/var/${SAMPLE}"
# Output files
TRIMMED_R1="${TRIMMED_DIR}/${SAMPLE}_R1_val_1.fq.gz"
TRIMMED_R2="${TRIMMED_DIR}/${SAMPLE}_R2_val_2.fq.gz"
ALIGNED_BAM="${ALIGNED_DIR}/${SAMPLE}.bam"
SORTED_BAM="${ALIGNED_DIR}/${SAMPLE}.sorted.bam"
DEDUP_BAM="${ALIGNED_DIR}/${SAMPLE}_marked_duplicates.bam"
RECAL_TABLE="${ALIGNED_DIR}/${SAMPLE}_recal_data.table"
RECAL_BAM="${ALIGNED_DIR}/${SAMPLE}_recalibrated.bam"
GVCF="${VAR_DIR}/${SAMPLE}.g.vcf.gz"
RAW_VCF="${VAR_DIR}/${SAMPLE}_raw_variants.vcf.gz"
METRICS="${ALIGNED_DIR}/${SAMPLE}_duplicate_metrics.txt"
# Check tools are available
echo "Checking required tools..."
echo "✓ FastQC:"
fastqc --version
echo "✓ Trim Galore:"
trim_galore --version
echo "✓ BWA:"
bwa || true # BWA doesn't have a --version flag, so we ignore the error
echo "✓ Samtools:"
samtools --version
echo "✓ GATK:"
gatk --version
echo "✓ R (for insert size histogram):"
R --version
echo "All required tools are available."
# Create output directories
mkdir -p ${QC_DIR} ${TRIMMED_DIR} ${ALIGNED_DIR} ${VAR_DIR}
echo "=========================================="
echo "GATK Variant Calling Pipeline - Complete"
echo "Sample: ${SAMPLE}"
echo "Based on NGS101 Best Practices"
echo "=========================================="
echo
# Step 1: Quality Control with FastQC
echo "[$(date)] Step 1: Running FastQC on raw reads..."
fastqc -o ${QC_DIR} -t ${THREADS} ${FASTQ_R1} ${FASTQ_R2}
echo "[$(date)] FastQC completed"
echo
# Step 2: Adapter Trimming and Quality Filtering
echo "[$(date)] Step 2: Adapter trimming with Trim Galore..."
trim_galore \
--paired \
--quality 20 \
--length 50 \
--fastqc \
--output_dir ${TRIMMED_DIR} \
${FASTQ_R1} ${FASTQ_R2}
echo "[$(date)] Adapter trimming completed"
echo
# Step 3: Read Alignment with BWA-MEM
echo "[$(date)] Step 3: Aligning reads with BWA-MEM..."
bwa mem \
-t ${THREADS} \
-M \
-R "@RG\tID:${SAMPLE}\tSM:${SAMPLE}\tPL:ILLUMINA\tLB:${SAMPLE}_lib" \
${REFERENCE} \
${TRIMMED_R1} ${TRIMMED_R2} \
| samtools view -Sb - > ${ALIGNED_BAM}
echo "[$(date)] Alignment completed"
echo
# Step 4: Sort and Index BAM file
echo "[$(date)] Step 4: Sorting BAM file..."
samtools sort -@ ${THREADS} -o ${SORTED_BAM} ${ALIGNED_BAM}
samtools index ${SORTED_BAM}
echo "[$(date)] Sorting completed"
echo
# Step 5: Mark Duplicates
echo "[$(date)] Step 5: Marking duplicates with GATK..."
gatk MarkDuplicates \
-I ${SORTED_BAM} \
-O ${DEDUP_BAM} \
-M ${METRICS}
echo "[$(date)] Mark duplicates completed"
echo
# Step 6: Base Quality Score Recalibration (BQSR) - Generate table
echo "[$(date)] Step 6: Generating BQSR recalibration table..."
gatk BaseRecalibrator \
-I ${DEDUP_BAM} \
-R ${REFERENCE} \
--known-sites ${DBSNP} \
--known-sites ${KNOWN_INDELS} \
-O ${RECAL_TABLE}
echo "[$(date)] BQSR table generated"
echo
# Step 7: Apply BQSR
echo "[$(date)] Step 7: Applying BQSR..."
gatk ApplyBQSR \
-I ${DEDUP_BAM} \
-R ${REFERENCE} \
--bqsr-recal-file ${RECAL_TABLE} \
-O ${RECAL_BAM}
echo "[$(date)] BQSR applied"
echo
# Step 8: Alignment Quality Assessment
echo "[$(date)] Step 8: Collecting alignment quality metrics..."
gatk CollectAlignmentSummaryMetrics \
-R ${REFERENCE} \
-I ${RECAL_BAM} \
-O ${QC_DIR}/${SAMPLE}_alignment_summary.txt
# Collect insert size metrics with histogram (requires R)
gatk CollectInsertSizeMetrics \
-I ${RECAL_BAM} \
-O ${QC_DIR}/${SAMPLE}_insert_size_metrics.txt \
-H ${QC_DIR}/${SAMPLE}_insert_size_histogram.pdf
echo "[$(date)] Alignment QC completed"
echo
# Step 9: Variant Calling with HaplotypeCaller (GVCF mode)
echo "[$(date)] Step 9: Calling variants with HaplotypeCaller in GVCF mode..."
gatk HaplotypeCaller \
-R ${REFERENCE} \
-I ${RECAL_BAM} \
-O ${GVCF} \
-ERC GVCF \
--dbsnp ${DBSNP}
echo "[$(date)] GVCF generation completed"
echo
# Step 10: Genotype GVCFs
echo "[$(date)] Step 10: Genotyping GVCF to VCF..."
gatk GenotypeGVCFs \
-R ${REFERENCE} \
-V ${GVCF} \
-O ${RAW_VCF}
echo "[$(date)] Genotyping completed"
echo
bash bash-gatk/gatk_pipeline.sh
The results output
── results
│ ├── aligned
│ │ └── sample1
│ │ ├── sample1.bam
│ │ ├── sample1_duplicate_metrics.txt
│ │ ├── sample1_marked_duplicates.bai
│ │ ├── sample1_marked_duplicates.bam
│ │ ├── sample1_recal_data.table
│ │ ├── sample1_recalibrated.bai
│ │ ├── sample1_recalibrated.bam
│ │ └── sample1.sorted.bam
│ ├── qc
│ │ └── sample1
│ │ ├── sample1_alignment_summary.txt
│ │ ├── sample1_insert_size_histogram.pdf
│ │ ├── sample1_insert_size_metrics.txt
│ │ ├── sample1_R1_fastqc.html
│ │ ├── sample1_R1_fastqc.zip
│ │ ├── sample1_R2_fastqc.html
│ │ └── sample1_R2_fastqc.zip
│ ├── trimmed
│ │ └── sample1
│ │ ├── sample1_R1.fastq.gz_trimming_report.txt
│ │ ├── sample1_R1_val_1_fastqc.html
│ │ ├── sample1_R1_val_1_fastqc.zip
│ │ ├── sample1_R1_val_1.fq.gz
│ │ ├── sample1_R2.fastq.gz_trimming_report.txt
│ │ ├── sample1_R2_val_2_fastqc.html
│ │ ├── sample1_R2_val_2_fastqc.zip
│ │ └── sample1_R2_val_2.fq.gz
│ └── var
│ └── sample1
│ ├── sample1.g.vcf.gz
│ ├── sample1.g.vcf.gz.tbi
│ ├── sample1_raw_variants.vcf.gz
│ └── sample1_raw_variants.vcf.gz.tbi
4.2 Validate
You can open the FastQC HTML reports in your browser to review the quality control results.
To inspect the variant calling output, run:
zcat results/var/sample1/sample1_raw_variants.vcf.gz
5. References
5.1 Primary Resources
- NGS101 GATK Tutorial: How to Analyze Whole Genome Sequencing Data - Complete walkthrough of GATK variant calling workflow
- GATK Best Practices: Broad Institute GATK Workflows - Official GATK germline variant calling guidelines
- nf-core Test Datasets: GitHub Repository - Publicly available test data for bioinformatics pipelines
5.2 Tools and Documentation
- Pixi Package Manager: Official Documentation - Modern, fast conda-based environment manager
- BWA Manual: BWA Homepage - Burrows-Wheeler Aligner documentation
- SAMtools: Official Site - Tools for manipulating SAM/BAM files
- GATK Documentation: Broad Institute - Comprehensive GATK tool documentation
- Trim Galore: Babraham Bioinformatics - Adapter trimming tool
- SnpEff: Official Site - Variant annotation and effect prediction
- bcftools: SAMtools bcftools - Variant calling and manipulation
5.3 Related Blog Posts
- Pixi - New conda era - Understanding modern package management with Pixi
- Containers on HPC: From Docker to Singularity and Apptainer - Container fundamentals for reproducible bioinformatics
- The Evolution of Version Control - Git's Role in Reproducible Bioinformatics (Part 1) - Version control basics for bioinformatics
5.4 Code Repository
- GitHub: variant-calling-gatk-pipeline-best-practice-from-scratch - Complete code from this blog post
5.5 Further Reading
- GATK Hard Filtering: GATK Forums Discussion - When to use hard filtering vs. VQSR
- GVCF Mode: Joint Genotyping Workflow - Understanding GVCF and joint calling
- Nextflow Documentation: Official Docs - Preview for Part 2
- nf-core: Best Practice Pipelines - Community-curated bioinformatics pipelines
Recap
While bash workflows with Pixi provide reproducible environments, they have limitations:
- Sequential execution (no parallel sample processing)
- Manual analysis management
- Limited scalability for local, cloud, or HPC deployment
Modern workflow management frameworks (e.g., WDL, Snakemake, Galaxy, Nextflow) address these challenges. In the next post, we'll transform this bash workflow into a Nextflow pipeline for improved automation and scalability.